Antiviral Antiviral Antivirals for Hepatitis B agents against human herpesviruses (HHVs) include acyclovir Acyclovir A guanosine analog that acts as an antimetabolite. Viruses are especially susceptible. Used especially against herpes. Herpes Zoster (Shingles), cidofovir, and foscarnet. Human herpesviruses are DNA viruses DNA Viruses Viruses whose nucleic acid is DNA. Virology in the Herpesviridae Herpesviridae A family of enveloped, linear, double-stranded DNA viruses infecting a wide variety of animals. Subfamilies, based on biological characteristics, include: alphaherpesvirinae; betaherpesvirinae; and gammaherpesvirinae. Herpes Simplex Virus 1 and 2 family. Herpes simplex Herpes Simplex A group of acute infections caused by herpes simplex virus type 1 or type 2 that is characterized by the development of one or more small fluid-filled vesicles with a raised erythematous base on the skin or mucous membrane. It occurs as a primary infection or recurs due to a reactivation of a latent infection. Congenital TORCH Infections virus Virus Viruses are infectious, obligate intracellular parasites composed of a nucleic acid core surrounded by a protein capsid. Viruses can be either naked (non-enveloped) or enveloped. The classification of viruses is complex and based on many factors, including type and structure of the nucleoid and capsid, the presence of an envelope, the replication cycle, and the host range. Virology ( HSV HSV Herpes simplex virus (HSV) is a double-stranded DNA virus belonging to the family Herpesviridae. Herpes simplex virus commonly causes recurrent infections involving the skin and mucosal surfaces, including the mouth, lips, eyes, and genitals. Herpes Simplex Virus 1 and 2), varicella-zoster virus Varicella-Zoster Virus Varicella-zoster virus (VZV) is a linear, double-stranded DNA virus in the Herpesviridae family. Varicella-zoster infections are highly contagious and transmitted through aerosolized respiratory droplets or contact with infected skin lesions. Varicella-Zoster Virus/Chickenpox (VZV), cytomegalovirus Cytomegalovirus CMV is a ubiquitous double-stranded DNA virus belonging to the Herpesviridae family. CMV infections can be transmitted in bodily fluids, such as blood, saliva, urine, semen, and breast milk. The initial infection is usually asymptomatic in the immunocompetent host, or it can present with symptoms of mononucleosis. Cytomegalovirus (CMV), Epstein-Barr virus Epstein-Barr Virus Epstein-Barr virus (EBV) is a linear, double-stranded DNA virus belonging to the Herpesviridae family. This highly prevalent virus is mostly transmitted through contact with oropharyngeal secretions from an infected individual. The virus can infect epithelial cells and B lymphocytes, where it can undergo lytic replication or latency. Epstein-Barr Virus ( EBV EBV Epstein-barr virus (EBV) is a linear, double-stranded DNA virus belonging to the herpesviridae family. This highly prevalent virus is mostly transmitted through contact with oropharyngeal secretions from an infected individual. The virus can infect epithelial cells and B lymphocytes, where it can undergo lytic replication or latency. Epstein-Barr Virus), and HHV-8 belong to the Herpesviridae Herpesviridae A family of enveloped, linear, double-stranded DNA viruses infecting a wide variety of animals. Subfamilies, based on biological characteristics, include: alphaherpesvirinae; betaherpesvirinae; and gammaherpesvirinae. Herpes Simplex Virus 1 and 2 family. Antivirals against the group generally act via inhibition of DNA DNA A deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine). DNA Types and Structure polymerase. Acyclovir Acyclovir A guanosine analog that acts as an antimetabolite. Viruses are especially susceptible. Used especially against herpes. Herpes Zoster (Shingles) (the prototypical nucleoside analog) requires viral kinase for phosphorylation Phosphorylation The introduction of a phosphoryl group into a compound through the formation of an ester bond between the compound and a phosphorus moiety. Post-translational Protein Processing to become a triphosphate, which is incorporated in viral DNA DNA A deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine). DNA Types and Structure. Cidofovir requires phosphorylation Phosphorylation The introduction of a phosphoryl group into a compound through the formation of an ester bond between the compound and a phosphorus moiety. Post-translational Protein Processing by host cellular kinase, which allows cidofovir to have activity against mutated viruses Viruses Minute infectious agents whose genomes are composed of DNA or RNA, but not both. They are characterized by a lack of independent metabolism and the inability to replicate outside living host cells. Virology and become deficient in viral kinase. Foscarnet (a pyrophosphate analog) does not require phosphorylation Phosphorylation The introduction of a phosphoryl group into a compound through the formation of an ester bond between the compound and a phosphorus moiety. Post-translational Protein Processing. Nephrotoxicity Nephrotoxicity Glycopeptides is a shared adverse effect in the agents. Acyclovir Acyclovir A guanosine analog that acts as an antimetabolite. Viruses are especially susceptible. Used especially against herpes. Herpes Zoster (Shingles) can also cause obstructive crystalline nephropathy and foscarnet carries a risk of electrolyte abnormalities and seizures Seizures A seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures. The nephrotoxic effect of cidofovir can be reduced with IV saline and probenecid Probenecid The prototypical uricosuric agent. It inhibits the renal excretion of organic anions and reduces tubular reabsorption of urate. Probenecid has also been used to treat patients with renal impairment, and, because it reduces the renal tubular excretion of other drugs, has been used as an adjunct to antibacterial therapy. Gout Drugs.
Last updated: Dec 15, 2025
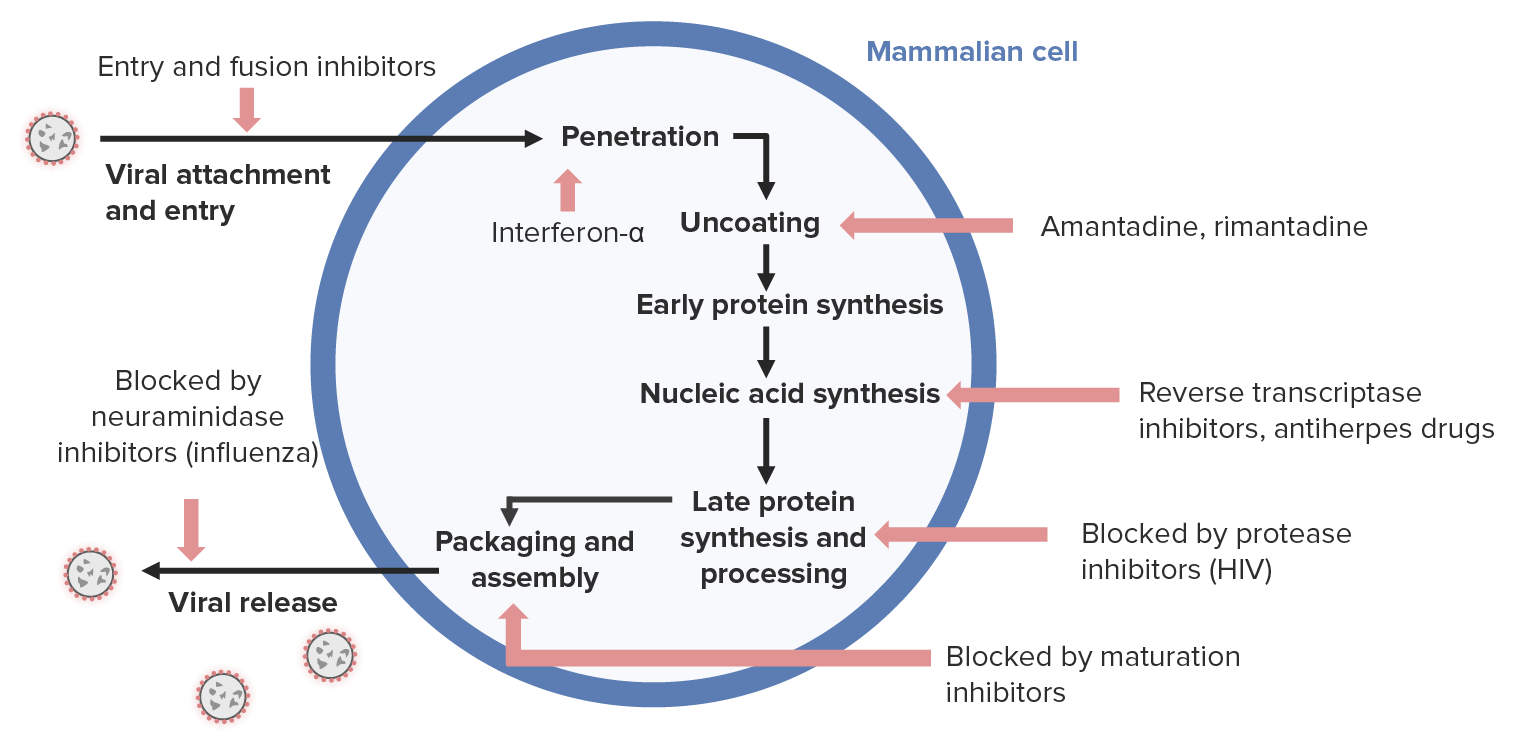
Antiviral drug mechanism:
Antivirals for herpesviruses (right side) generally inhibit nucleic acid synthesis.
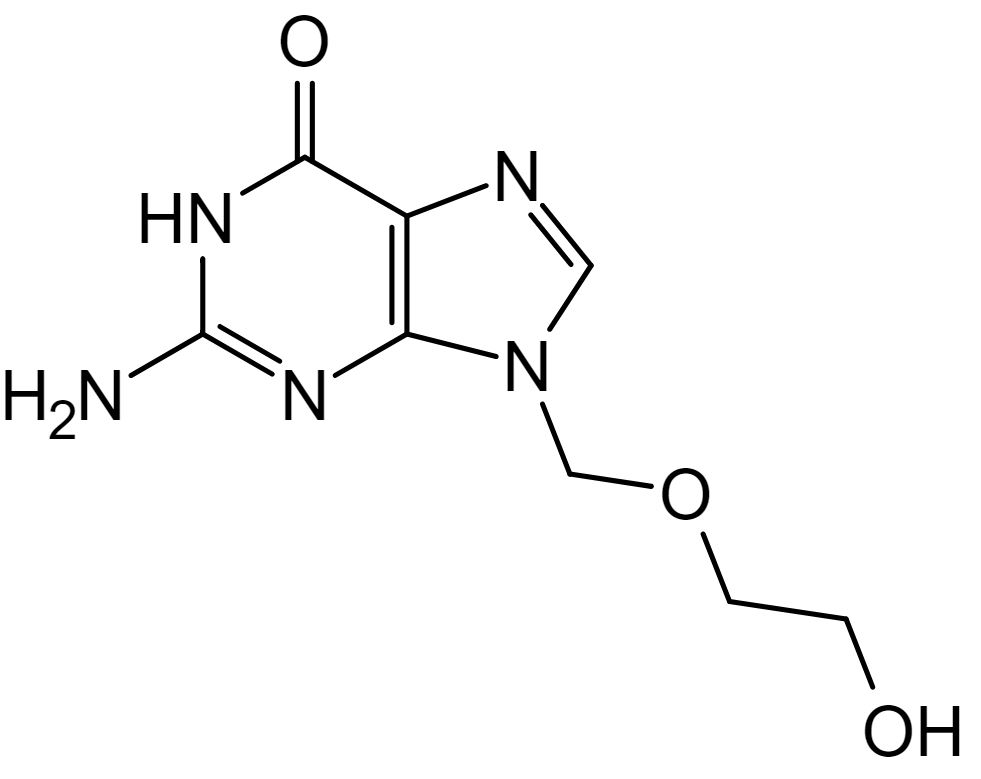
Chemical structure of acyclovir
Image: “Aciclovir structure” by Steffen 962. License: Public DomainAcyclovir Acyclovir A guanosine analog that acts as an antimetabolite. Viruses are especially susceptible. Used especially against herpes. Herpes Zoster (Shingles):

Mechanism of action of acyclovir:
1) Acyclovir undergoes sequential phosphorylation, which is facilitated by herpesvirus thymidine kinase (TK) and host kinases. Since viral TK is required for phosphorylation of acyclovir, only cells infected with the virus are affected by the drug.
2) Host enzymes convert the monophosphate to acyclovir triphosphate, which subsequently competes with deoxyguanosine triphosphate (dGTP) for the viral DNA polymerase.
3) Acyclovir is added to the growing strand during viral replication instead of guanosine triphosphate (GTP), which stops viral replication by halting the elongation of the DNA molecule.
Acyclovir Acyclovir A guanosine analog that acts as an antimetabolite. Viruses are especially susceptible. Used especially against herpes. Herpes Zoster (Shingles) (and other related agents):
Acyclovir Acyclovir A guanosine analog that acts as an antimetabolite. Viruses are especially susceptible. Used especially against herpes. Herpes Zoster (Shingles):
| Agent | Characteristics and mechanism of action | Pharmacokinetics Pharmacokinetics Pharmacokinetics is the science that analyzes how the human body interacts with a drug. Pharmacokinetics examines how the drug is absorbed, distributed, metabolized, and excreted by the body. Pharmacokinetics and Pharmacodynamics | Approved indication | Adverse drug effects |
|---|---|---|---|---|
| Acyclovir Acyclovir A guanosine analog that acts as an antimetabolite. Viruses are especially susceptible. Used especially against herpes. Herpes Zoster (Shingles) |
|
Poor oral bioavailability Bioavailability Pharmacokinetics and Pharmacodynamics |
|
|
| Valacyclovir Valacyclovir A prodrug of acyclovir that is used in the treatment of herpes zoster and herpes simplex virus infection of the skin and mucous membranes, including genital herpes. Herpes Zoster (Shingles) |
|
Good oral bioavailability Bioavailability Pharmacokinetics and Pharmacodynamics | ||
| Penciclovir |
|
Topical | HSV HSV Herpes simplex virus (HSV) is a double-stranded DNA virus belonging to the family Herpesviridae. Herpes simplex virus commonly causes recurrent infections involving the skin and mucosal surfaces, including the mouth, lips, eyes, and genitals. Herpes Simplex Virus 1 and 2 ( herpes labialis Herpes labialis Herpes simplex, caused by type 1 virus, primarily spread by oral secretions and usually occurring as a concomitant of fever. It may also develop in the absence of fever or prior illness. It commonly involves the facial region, especially the lips and the nares. Herpes Simplex Virus 1 and 2) | Mild side effects |
| Famciclovir |
|
Better oral bioavailability Bioavailability Pharmacokinetics and Pharmacodynamics than penciclovir |
|
|
| Ganciclovir |
|
|
Cytomegalovirus Cytomegalovirus CMV is a ubiquitous double-stranded DNA virus belonging to the Herpesviridae family. CMV infections can be transmitted in bodily fluids, such as blood, saliva, urine, semen, and breast milk. The initial infection is usually asymptomatic in the immunocompetent host, or it can present with symptoms of mononucleosis. Cytomegalovirus (CMV) infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease |
|
| Valganciclovir |
|
Better oral bioavailability Bioavailability Pharmacokinetics and Pharmacodynamics than ganciclovir |
Cidofovir is a cytidine nucleotide analog.
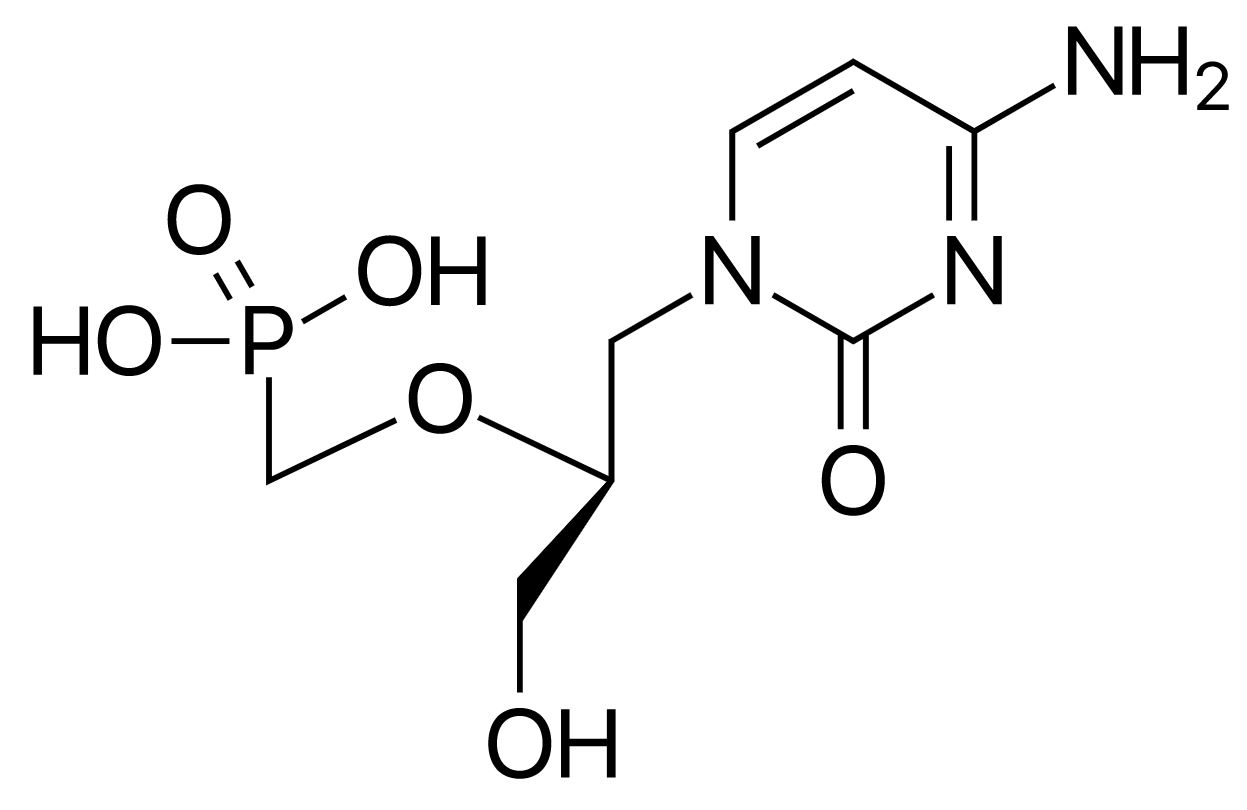
Chemical structure of cidofovir
Image: “Cidofovir” by ljfa-ag. License: Public DomainFoscarnet is a pyrophosphate analog.
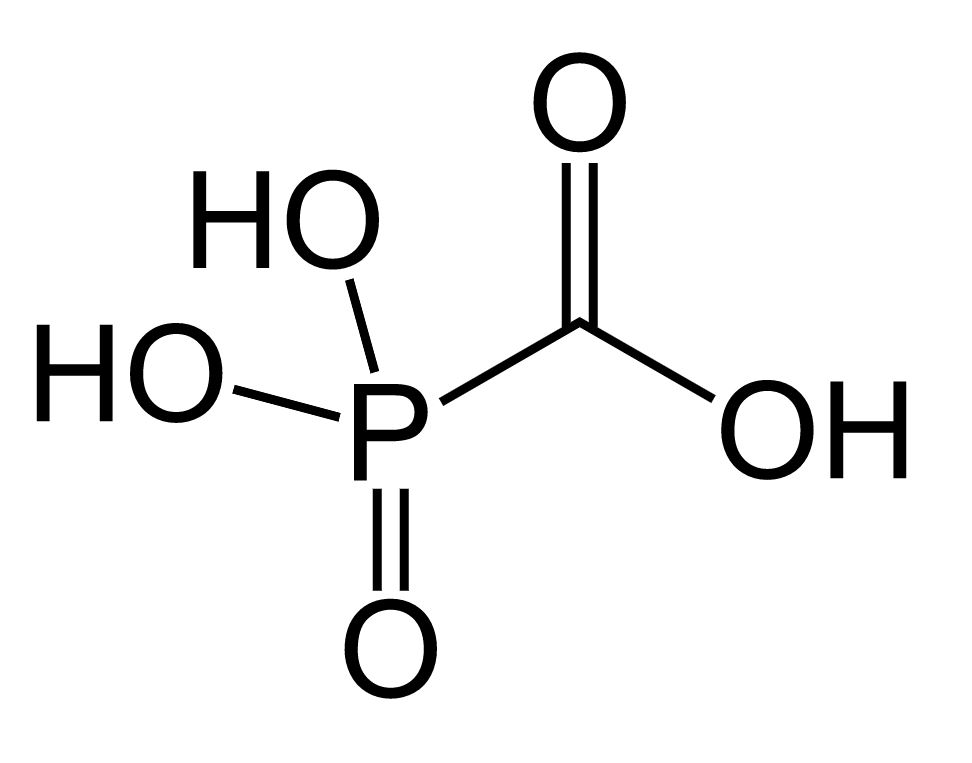
Chemical structure of foscarnet
Image: “Foscarnet” by Fvasconcellos. License: Public Domain| Antiviral Antiviral Antivirals for Hepatitis B agent | Mechanism of action | Indications | Major adverse effects |
|---|---|---|---|
| Acyclovir Acyclovir A guanosine analog that acts as an antimetabolite. Viruses are especially susceptible. Used especially against herpes. Herpes Zoster (Shingles) (nucleoside analog) | Polymerase inhibitor requiring phosphorylation Phosphorylation The introduction of a phosphoryl group into a compound through the formation of an ester bond between the compound and a phosphorus moiety. Post-translational Protein Processing by viral kinase |
|
Acute kidney injury Acute Kidney Injury Acute kidney injury refers to sudden and often reversible loss of renal function, which develops over days or weeks. Azotemia refers to elevated levels of nitrogen-containing substances in the blood that accompany AKI, which include BUN and creatinine. Acute Kidney Injury (obstructive nephropathy) |
| Cidofovir (nucleotide analog) | Polymerase inhibitor requiring phosphorylation Phosphorylation The introduction of a phosphoryl group into a compound through the formation of an ester bond between the compound and a phosphorus moiety. Post-translational Protein Processing by host kinase (not viral kinase) | Approved for CMV retinitis CMV retinitis Infection of the retina by cytomegalovirus characterized by retinal necrosis, hemorrhage, vessel sheathing, and retinal edema. Cytomegalovirus retinitis is a major opportunistic infection in AIDS patients and can cause blindness. Retinal Detachment with activity against other viruses Viruses Minute infectious agents whose genomes are composed of DNA or RNA, but not both. They are characterized by a lack of independent metabolism and the inability to replicate outside living host cells. Virology* | Nephrotoxicity Nephrotoxicity Glycopeptides (risk decreased by IV saline and probenecid Probenecid The prototypical uricosuric agent. It inhibits the renal excretion of organic anions and reduces tubular reabsorption of urate. Probenecid has also been used to treat patients with renal impairment, and, because it reduces the renal tubular excretion of other drugs, has been used as an adjunct to antibacterial therapy. Gout Drugs) |
| Foscarnet (pyrophosphate analog) | Polymerase inhibitor not requiring phosphorylation Phosphorylation The introduction of a phosphoryl group into a compound through the formation of an ester bond between the compound and a phosphorus moiety. Post-translational Protein Processing | Resistant CMV and HSV HSV Herpes simplex virus (HSV) is a double-stranded DNA virus belonging to the family Herpesviridae. Herpes simplex virus commonly causes recurrent infections involving the skin and mucosal surfaces, including the mouth, lips, eyes, and genitals. Herpes Simplex Virus 1 and 2 |
|