Marfan syndrome is a genetic condition with autosomal dominant inheritance Autosomal dominant inheritance Autosomal Recessive and Autosomal Dominant Inheritance. Marfan syndrome affects the elasticity Elasticity Resistance and recovery from distortion of shape. Skeletal Muscle Contraction of connective tissues throughout the body, most notably in the cardiovascular, ocular, and musculoskeletal systems. The skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions, lungs Lungs Lungs are the main organs of the respiratory system. Lungs are paired viscera located in the thoracic cavity and are composed of spongy tissue. The primary function of the lungs is to oxygenate blood and eliminate CO2. Lungs: Anatomy, and central nervous system Central nervous system The main information-processing organs of the nervous system, consisting of the brain, spinal cord, and meninges. Nervous System: Anatomy, Structure, and Classification are also affected. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship are usually tall with long limbs, fingers, and toes, and hypermobile joints. Associated conditions include aortic aneurysm Aortic aneurysm An abnormal balloon- or sac-like dilatation in the wall of aorta. Thoracic Aortic Aneurysms or dissection, mitral valve Mitral valve The valve between the left atrium and left ventricle of the heart. Heart: Anatomy prolapse, and lens Lens A transparent, biconvex structure of the eye, enclosed in a capsule and situated behind the iris and in front of the vitreous humor (vitreous body). It is slightly overlapped at its margin by the ciliary processes. Adaptation by the ciliary body is crucial for ocular accommodation. Eye: Anatomy dislocation. Diagnosis is made clinically with set criteria, and genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies is done only when it may affect the management. Medical or surgical management is based on clinical manifestations. Cardiovascular involvement is followed closely, as it is the main cause of mortality Mortality All deaths reported in a given population. Measures of Health Status.
Last updated: Dec 15, 2025

Family members with Marfan’s syndrome
Image: “Family members with Marfan’s syndrome” by Birjand Atherosclerosis and Coronary Artery Research Centre, Birjand University of Medical Sciences, Birjand, Iran. License: CC BY 3.0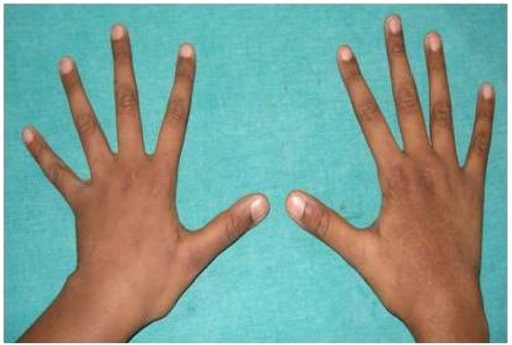
Photograph showing arachnodactyly
Image: “Arachnodactyly” by Department of Pedodontics, SRM Dental College, SRM University, Chennai 600078, India. License: CC BY 3.0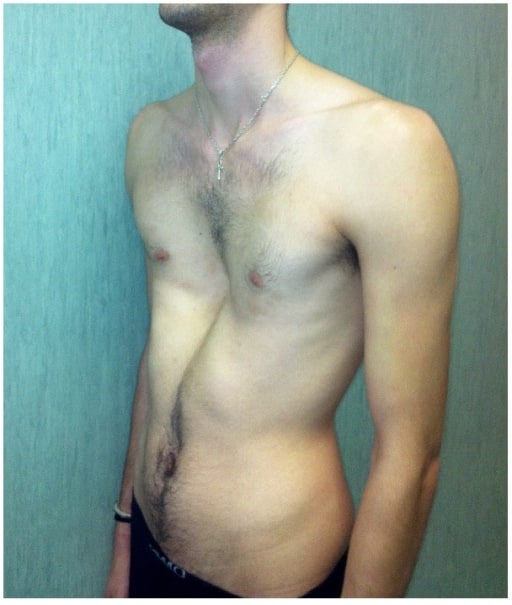
Severe pectus excavatum in a man affected by Marfan’s syndrome
Image: “fig2” by Fernando De Maio et al. License: CC BY 4.0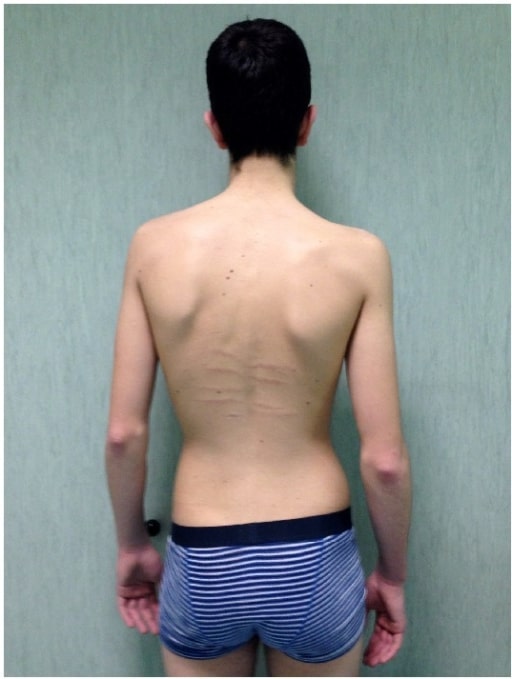
A man affected by Marfan’s syndrome with scoliosis in addition to dorsal skin striae and reduced elbow extension
Image: “fig5” by Fernando De Maio et al. License: CC BY 4.0
A patient with Marfan syndrome with an associated wrist and thumb sign:
(a) The “wrist sign” is positive when the thumb overlaps the fifth finger when grasping the contralateral wrist.
(b) The “thumb sign” is positive when the thumb extends well beyond the ulnar border of the hand when overlapped by the fingers.
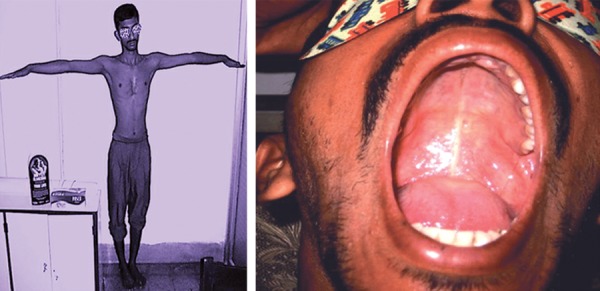
An arm span greater than the patient’s height, and a high-arched palate
Image: “Arm span more than height and high arched palate” by Consultant, Eye and Glaucoma Care, Gariahat, Kolkata-700029, West Bengal, India. License: CC BY 3.0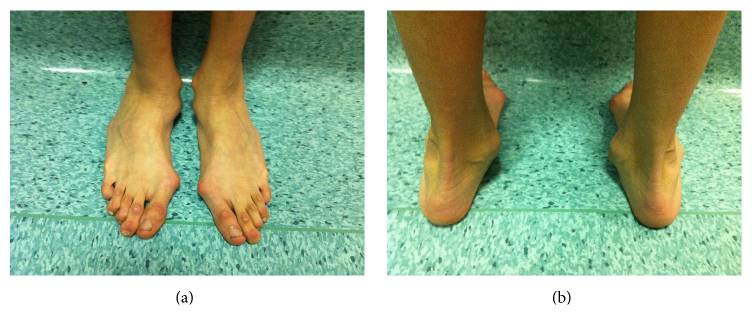
Hindfoot deformity with a marked valgus heel in a man affected by Marfan’s syndrome
Image: “Hindfoot deformity” by Fernando De Maio et al. License: CC BY 4.0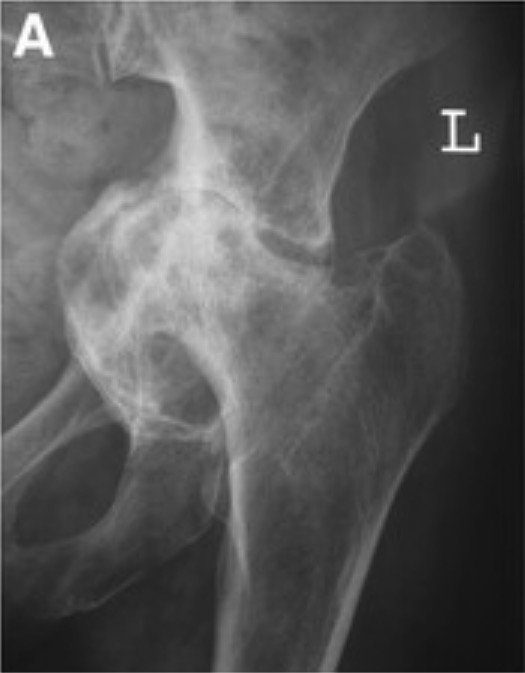
Radiograph demonstrating protrusio acetabuli: Notice that the femoral head and socket protrude into the pelvis.
Image: “Grade III protrusio acetabuli” by Orthopaedic Research Fellow, Royal Infirmary of Edinburgh, Little France EH16 4SA, UK. License: CC BY 2.0, edited by Lecturio.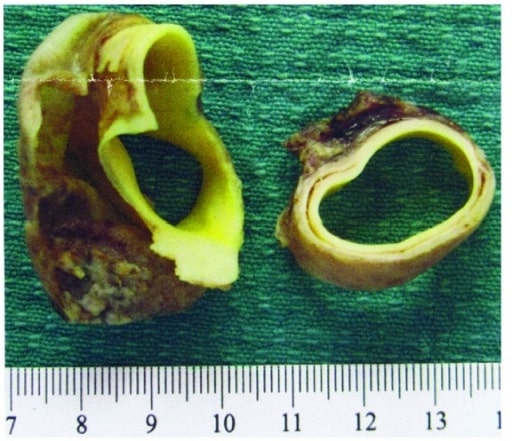
Gross pathology specimen of an aortic dissection flap and aortic wall
Image: “Aortic dissection flap and aortic wall” by Department of Obstetrics and Gynaecology, Women’s and Children’s Hospital, Adelaide, SA 5006, Australia. License: CC BY 3.0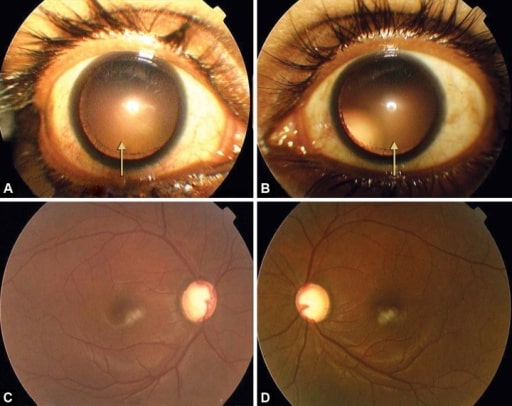
A and B: symmetrical temporal and upward subluxation of the lens on dilation
C and D: advanced glaucomatous cupping in both eyes
| Wrist and thumb sign |
|
| Pectus carinatum Pectus carinatum A developmental anomaly characterized by abnormal anterior protrusion of the sternum and adjacent costal cartilage. Cardiovascular Examination deformity Deformity Examination of the Upper Limbs | 2 points |
| Pectus excavatum Pectus Excavatum Cardiovascular Examination or chest asymmetry Asymmetry Examination of the Upper Limbs | 1 point |
| Hindfoot deformity Deformity Examination of the Upper Limbs | 2 points |
| Plain pes planus Pes Planus Ehlers-Danlos Syndrome | 1 point |
| Pneumothorax Pneumothorax A pneumothorax is a life-threatening condition in which air collects in the pleural space, causing partial or full collapse of the lung. A pneumothorax can be traumatic or spontaneous. Patients present with a sudden onset of sharp chest pain, dyspnea, and diminished breath sounds on exam. Pneumothorax | 2 points |
| Dural ectasia | 2 points |
| Protrusio acetabuli | 2 points |
| ↓ US/LS and ↑ arm Arm The arm, or “upper arm” in common usage, is the region of the upper limb that extends from the shoulder to the elbow joint and connects inferiorly to the forearm through the cubital fossa. It is divided into 2 fascial compartments (anterior and posterior). Arm: Anatomy span/height and no severe scoliosis Scoliosis Scoliosis is a structural alteration of the vertebral column characterized by a lateral spinal curvature of greater than 10 degrees in the coronal plane. Scoliosis can be classified as idiopathic (in most cases) or secondary to underlying conditions. Scoliosis | 1 point |
| Scoliosis Scoliosis Scoliosis is a structural alteration of the vertebral column characterized by a lateral spinal curvature of greater than 10 degrees in the coronal plane. Scoliosis can be classified as idiopathic (in most cases) or secondary to underlying conditions. Scoliosis or kyphosis Kyphosis Deformities of the spine characterized by an exaggerated convexity of the vertebral column. The forward bending of the thoracic region usually is more than 40 degrees. This deformity sometimes is called round back or hunchback. Osteoporosis | 1 point |
| ↓ elbow extension Extension Examination of the Upper Limbs | 1 point |
Facial features (at least 3 of 5)
|
1 point |
| Skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions striae | 1 point |
| Myopia Myopia Refractive Errors | 1 point |
| Mitral valve Mitral valve The valve between the left atrium and left ventricle of the heart. Heart: Anatomy prolapse | 1 point |
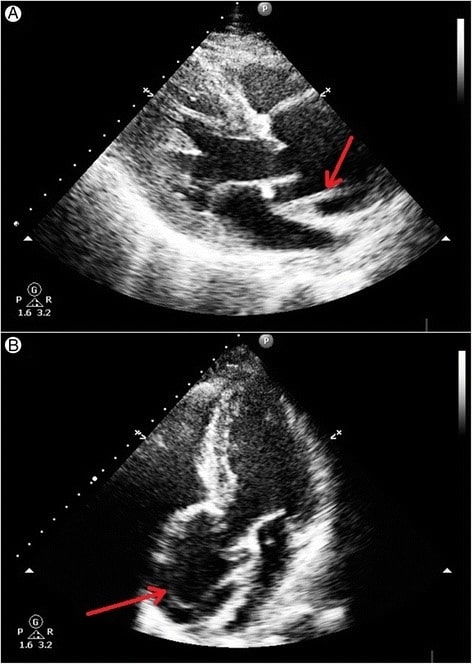
Transthoracic echocardiography showing an intimal tear in the dilated aortic root above the aortic valve level:
A: Linear intimal tear is seen just above the aortic valve in systole.
B: Linear intimal flap is visible in the dilated aortic root.
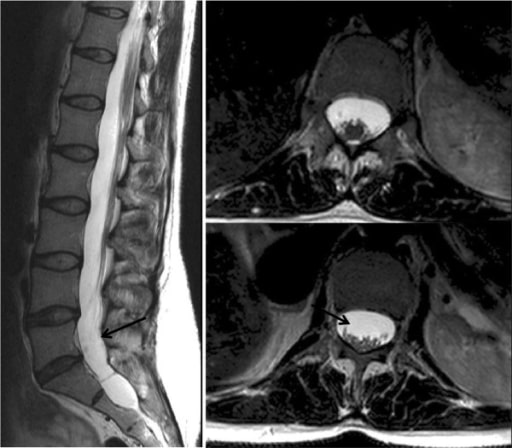
Magnetic resonance imaging demonstrating dural ectasia: This is ballooning or widening of the dural sac; it may be associated with herniation of the nerve root sleeves through their associated foramina (see arrows).
Image: “CMR” by Manchester Heart Centre, Manchester Royal Infirmary, Oxford Road, Manchester M13 9WL, UK. License: CC BY 2.0The management of MFS is based on the clinical manifestations. A multidisciplinary team of cardiologists, ophthalmologists, orthopedists, and cardiovascular surgeons is needed.
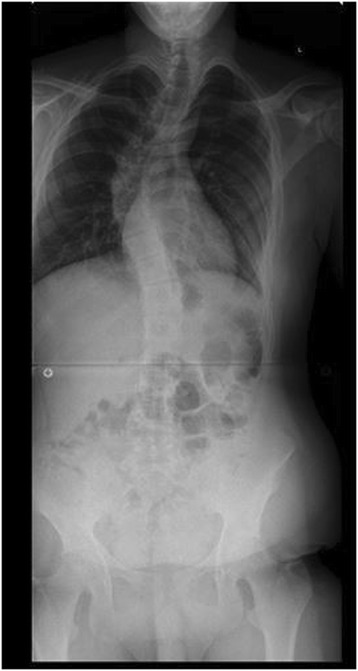
Pre-operative radiograph of a teenage boy with Marfan syndrome and scoliosis
Image: “Figure 1” by Levy et al. License: CC BY 4.0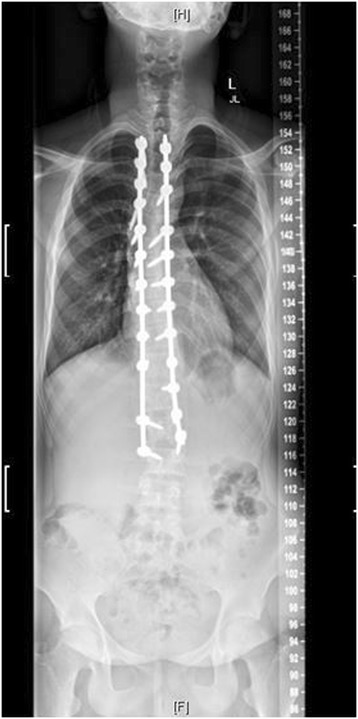
Post-operative radiograph of the same patient
Image: “Figure 4” by Levy et al. License: CC BY 4.0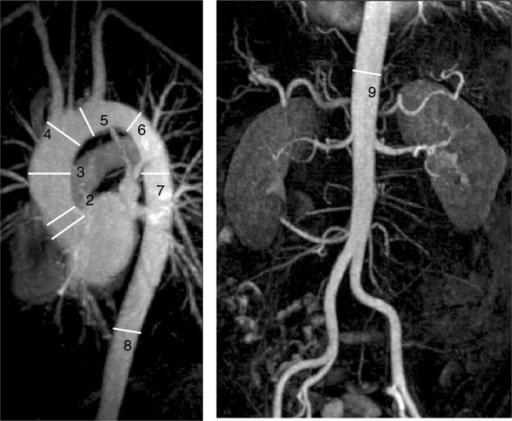
Cardiac MRI of the aorta demonstrating the standard imaging landmarks for the thoracoabdominal aorta used for monitoring: This image demonstrates dilation of the mid-ascending aorta (3).
Image: “F11” by Manchester Heart Centre, Manchester Royal Infirmary, Oxford Road, Manchester M13 9WL, UK. License: CC BY 2.0