Cholesterol is an important lipid molecule that is used for many biologic functions. Cholesterol can either be synthesized from endogenous acetyl-CoA Acetyl-CoA Acetyl CoA participates in the biosynthesis of fatty acids and sterols, in the oxidation of fatty acids and in the metabolism of many amino acids. It also acts as a biological acetylating agent. Citric Acid Cycle or absorbed from food in the GI tract. Because cholesterol is lipophilic, it must be transported through the bloodstream via lipoproteins Lipoproteins Lipid-protein complexes involved in the transportation and metabolism of lipids in the body. They are spherical particles consisting of a hydrophobic core of triglycerides and cholesterol esters surrounded by a layer of hydrophilic free cholesterol; phospholipids; and apolipoproteins. Lipoproteins are classified by their varying buoyant density and sizes. Lipid Metabolism, where it can be picked up by hepatocytes Hepatocytes The main structural component of the liver. They are specialized epithelial cells that are organized into interconnected plates called lobules. Liver: Anatomy or peripheral tissues. There, cholesterol can be stored, used in cellular membranes, or used as a precursor for steroid hormones Steroid hormones Steroid hormones produced by the gonads. They stimulate reproductive organs, germ cell maturation, and the secondary sex characteristics in the males and the females. The major sex steroid hormones include estradiol; progesterone; and testosterone. Hormones: Overview and Types. The human body cannot degrade cholesterol’s ring structure, so the only mechanism for potential excretion is through the production of bile Bile An emulsifying agent produced in the liver and secreted into the duodenum. Its composition includes bile acids and salts; cholesterol; and electrolytes. It aids digestion of fats in the duodenum. Gallbladder and Biliary Tract: Anatomy acids Acids Chemical compounds which yield hydrogen ions or protons when dissolved in water, whose hydrogen can be replaced by metals or basic radicals, or which react with bases to form salts and water (neutralization). An extension of the term includes substances dissolved in media other than water. Acid-Base Balance.
Last updated: Apr 25, 2025
Cholesterol’s synthesis Synthesis Polymerase Chain Reaction (PCR) pathway overlaps with the synthesis Synthesis Polymerase Chain Reaction (PCR) of ketone bodies Ketone bodies The metabolic substances acetone; 3-hydroxybutyric acid; and acetoacetic acid (acetoacetates). They are produced in the liver and kidney during fatty acids oxidation and used as a source of energy by the heart, muscle and brain. Ketone Body Metabolism. The steps of this process include:
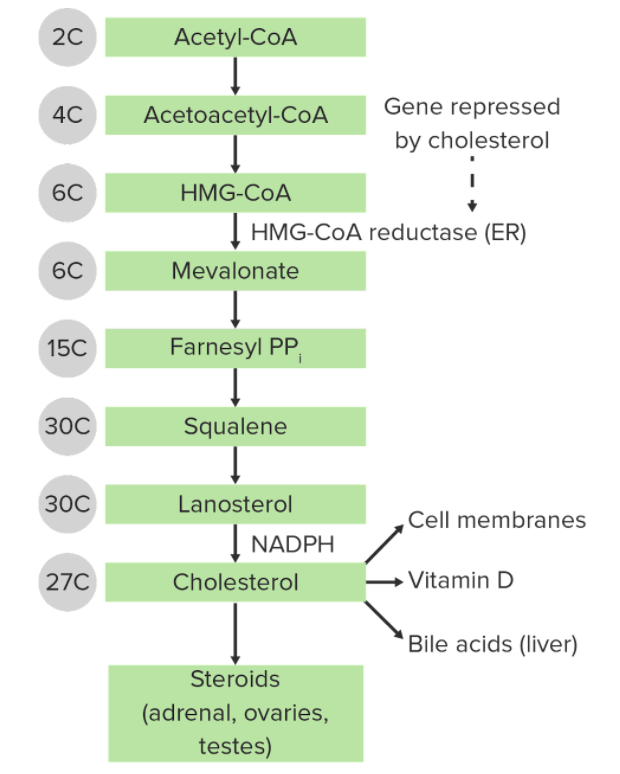
The synthesis of cholesterol by the mevalonate pathway:
The number of carbons within each chemical structure is noted to the left.
ER: endoplasmic reticulum
HMG-CoA: 3-hydroxy-3-methyl-glutaryl-coenzyme A
PP: pyrophosphatase
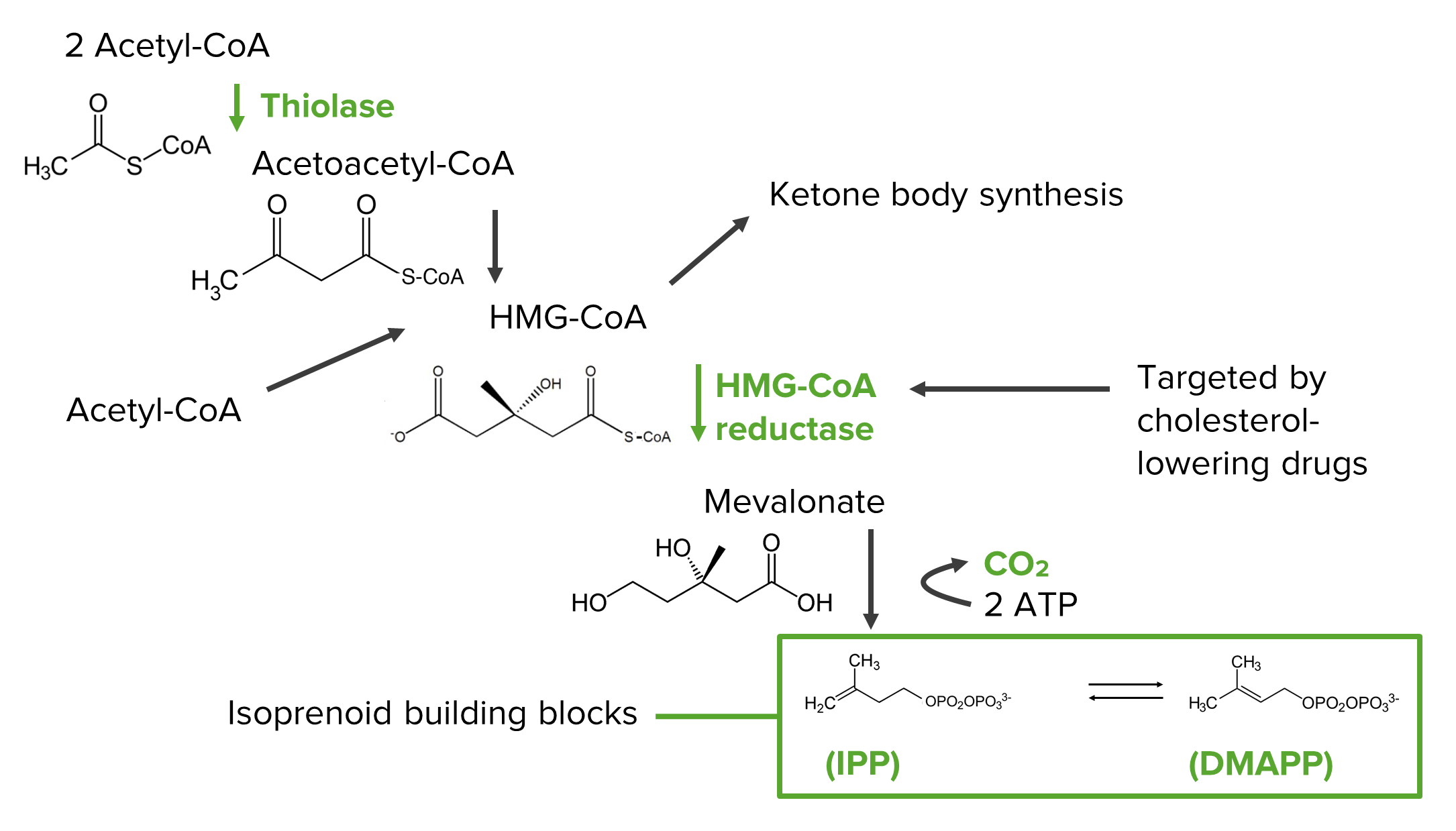
Diagram showing the steps of the mevalonate pathway in the synthesis of the isoprenoid precursors, which are the building blocks of cholesterol:
HMG-CoA: 3-hydroxy-3-methyl-glutaryl-CoA
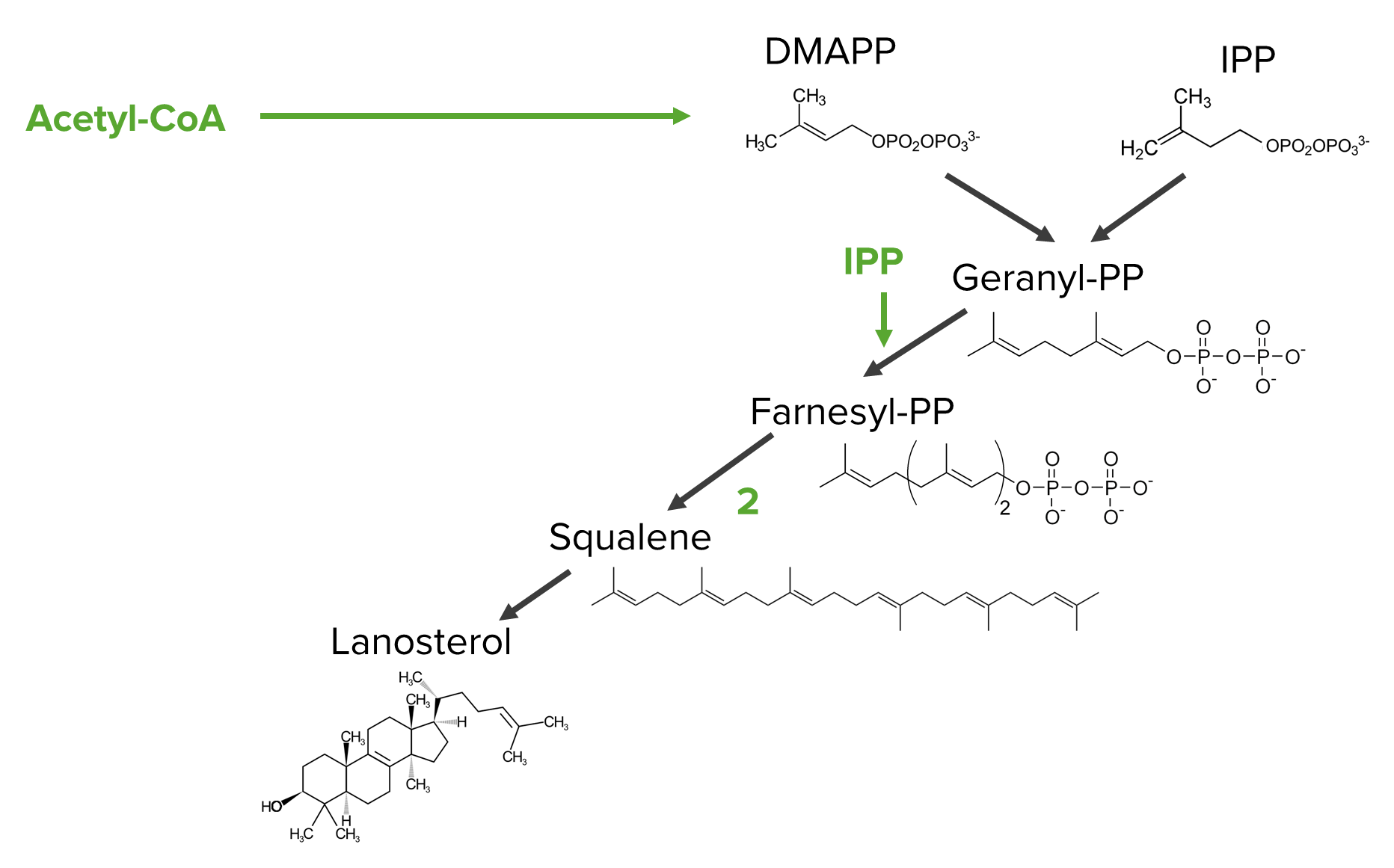
Diagram showing the steps of the mevalonate pathway from the isoprenoid precursors to lanosterol
Image by Lecturio.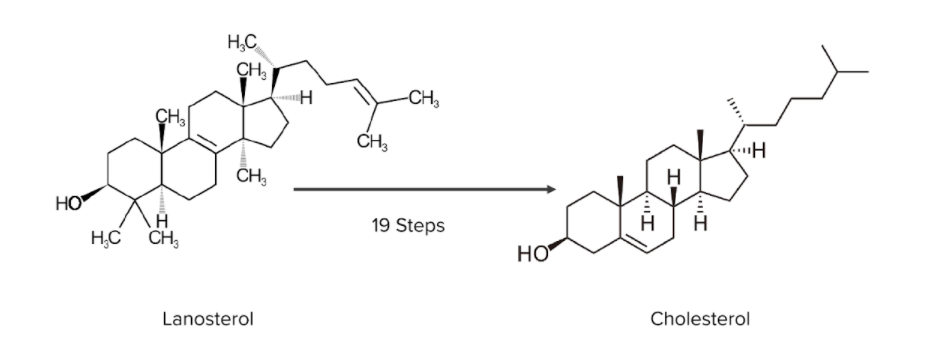
Diagram numbering the rest of the steps needed for the transformation of lanosterol into cholesterol:
The image provides a good comparison between the 2 molecules. Note how the cyclical structures are maintained throughout the conversion process.
Cholesterol is a:
| Lipoprotein | Cholesterol/ cholesterol ester Cholesterol Ester Fatty acid esters of cholesterol which constitute about two-thirds of the cholesterol in the plasma. The accumulation of cholesterol esters in the arterial intima is a characteristic feature of atherosclerosis. Lipid Metabolism |
Triglyceride | Protein | Phospholipid |
|---|---|---|---|---|
| Chylomicrons | 1%/3% | 85% | 2% | 8% |
| VLDL | 7%/10% | 55% | 9% | 20% |
| IDL | 8%/30% | 26% | 11% | 22% |
| LDL | 10%/35% | 10% | 20% | 20% |
| HDL | 4%/12% | 5% | 45% | 25% |
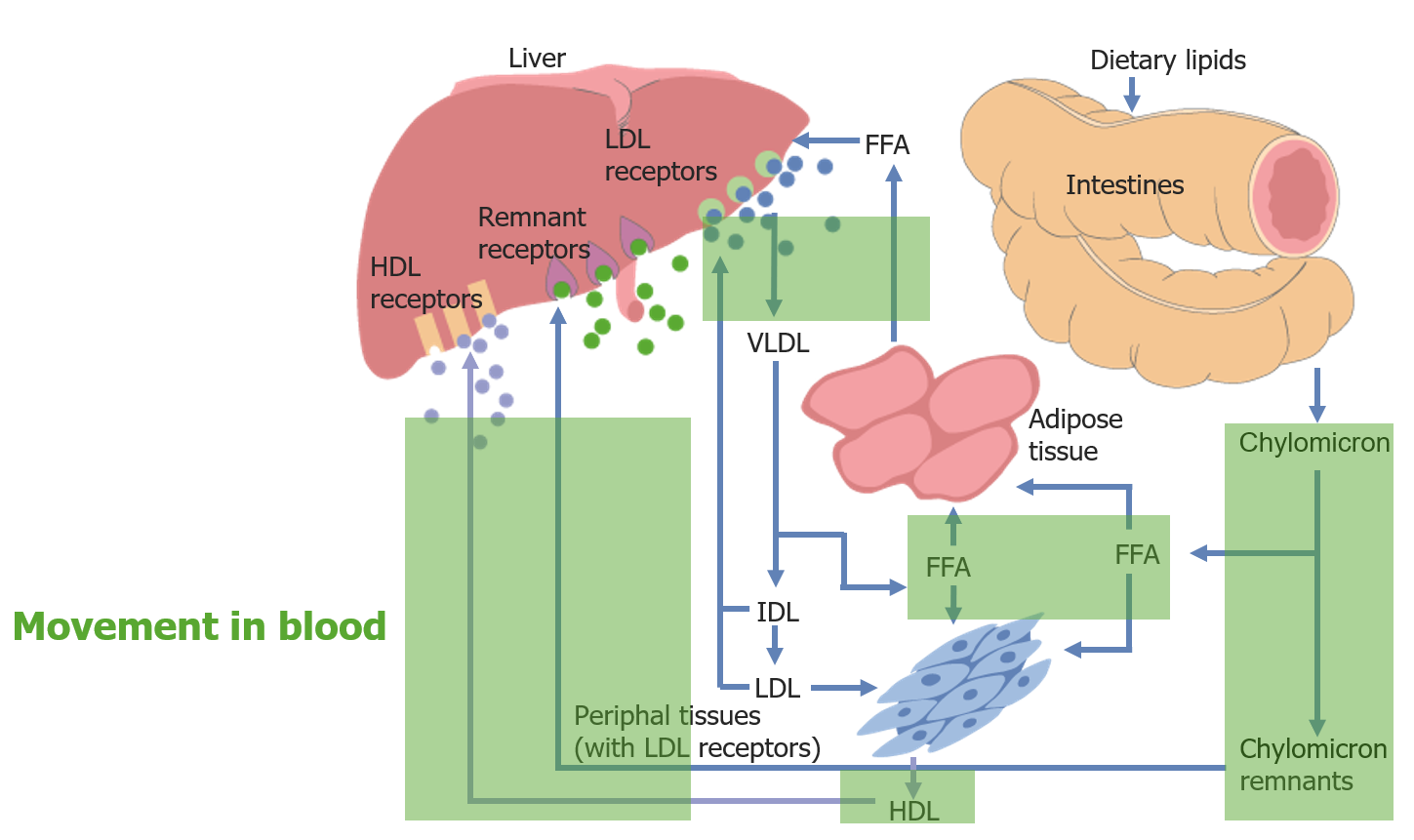
Movement of cholesterol throughout the body:
Movement in the blood is highlighted in green.
FFA: free fatty acid
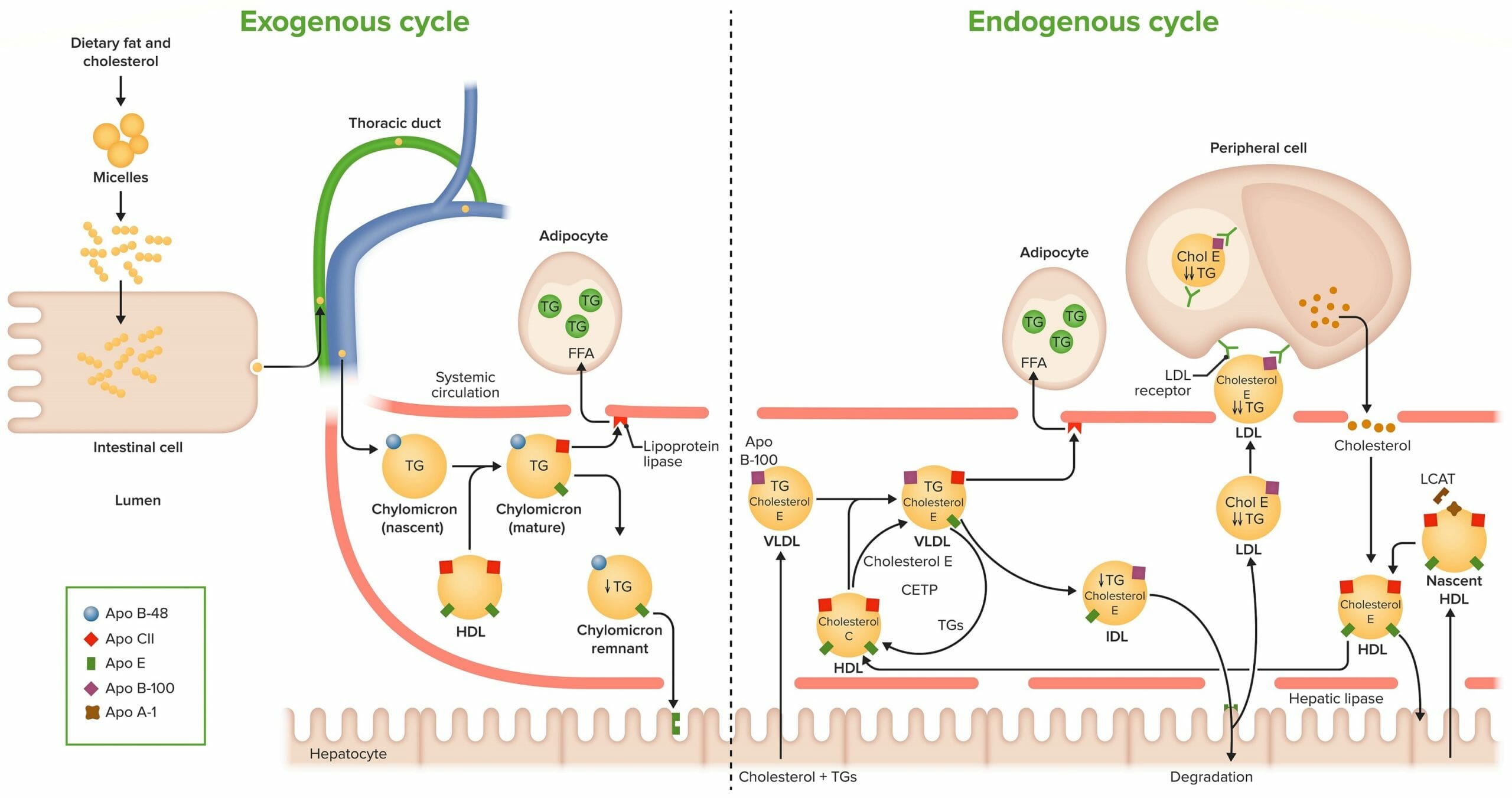
Exogenous cycle of cholesterol (Chol) transport and uptake:
Dietary fat and cholesterol are metabolized in the intestinal lumen to form micelles and are absorbed from the intestinal wall. In the intestinal cell, these lipids are coated by phospholipids, similar to that of the cell wall. Apolipoprotein (Apo) B-48 is attached and is necessary for release. The particle is now called a chylomicron, and it is secreted in the lymphatics, from which it enters systemic circulation. Chylomicrons take Apo C-II and Apo E from HDL. Apo C-II activates lipoprotein lipase (LPL), which degrades triglycerides (TGs). Apo E is used for the uptake of chylomicron remnants by the hepatocytes.
Endogenous cycle of cholesterol transport and uptake:
The liver synthesizes very-low-density lipoprotein (VLDL), which contains Apo B-100. In the circulation, Apo C-II and Apo E are exchanged between VLDL and high-density lipoprotein (HDL). Apo C-II activates LPL, which degrades TGs. In the circulation cholesteryl ester transfer protein (CETP) facilitates the exchange of TGs and cholesteryl esters between HDL and VLDL. The remnant of VLDL is called intermediate-density lipoprotein (IDL). It is taken up by the liver, where it is either degraded or released as LDL. It contains Apo B-100, which binds to the LDL receptor in the peripheral tissues and facilitates its uptake. HDL are particles formed in blood. They contain Apo A-I, which is made by hepatocytes or enteric cells. Nascent HDL contains only apolipoproteins. It takes up cholesterol from nonhepatic (peripheral tissues). To trap cholesterol inside HDL, the blood enzyme lecithin-cholesterol acetyltransferase (LCAT) facilitates its esterification. LCAT is synthesized and secreted by the liver. HDL returns cholesterol esters to the liver.
FFA: free fatty acid
Mechanism:
Regulation:
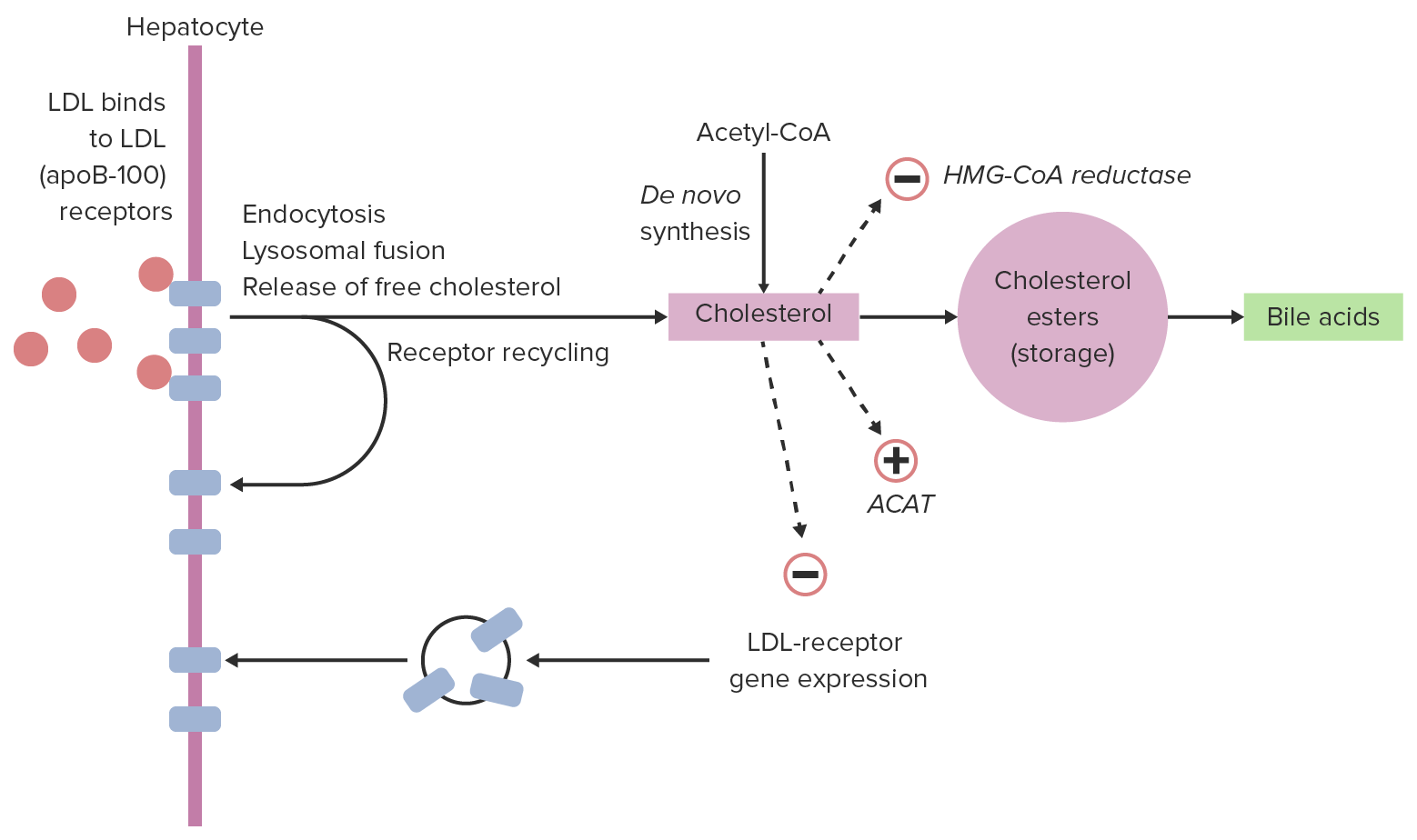
Cellular uptake of LDL and regulation:
Within a hepatocyte, LDL can bind with LDL receptors, allowing for endocytosis. LDL is released from the receptor, which returns to the cell surface. The LDL particle is broken down, releasing cholesterol. This cholesterol can down-regulate expression of LDL receptors and inhibit further cholesterol synthesis and can then either be stored or used (in this case, converted to bile acids).
ACAT: acyl cholesterol acyltransferase, which converts cholesterol to cholesterol esters
HMG-CoA: 3-hydroxy-3-methyl-glutaryl-coenzyme A
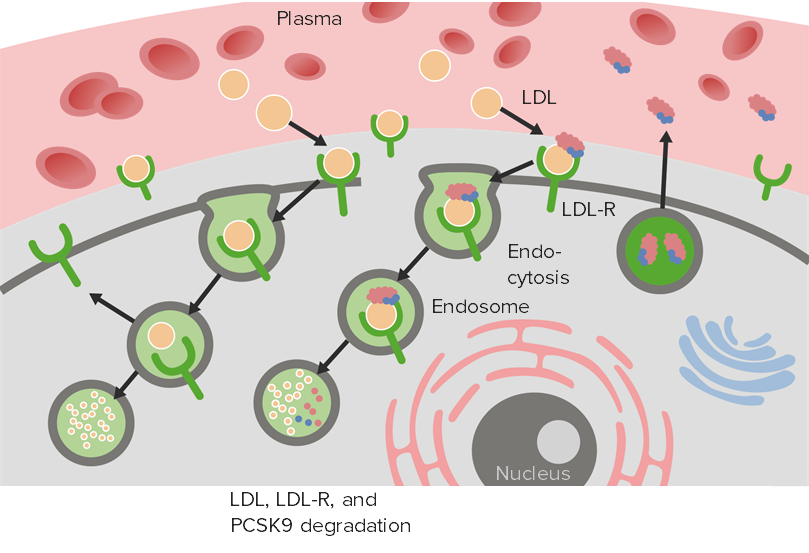
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is an enzyme secreted by the liver into the bloodstream that binds to LDL receptors (both inside and outside the hepatocyte) and subsequently promotes LDL receptor endocytosis for degradation.
LDL-R: LDL receptor

Chemical structures of bile acids
Image by Lecturio.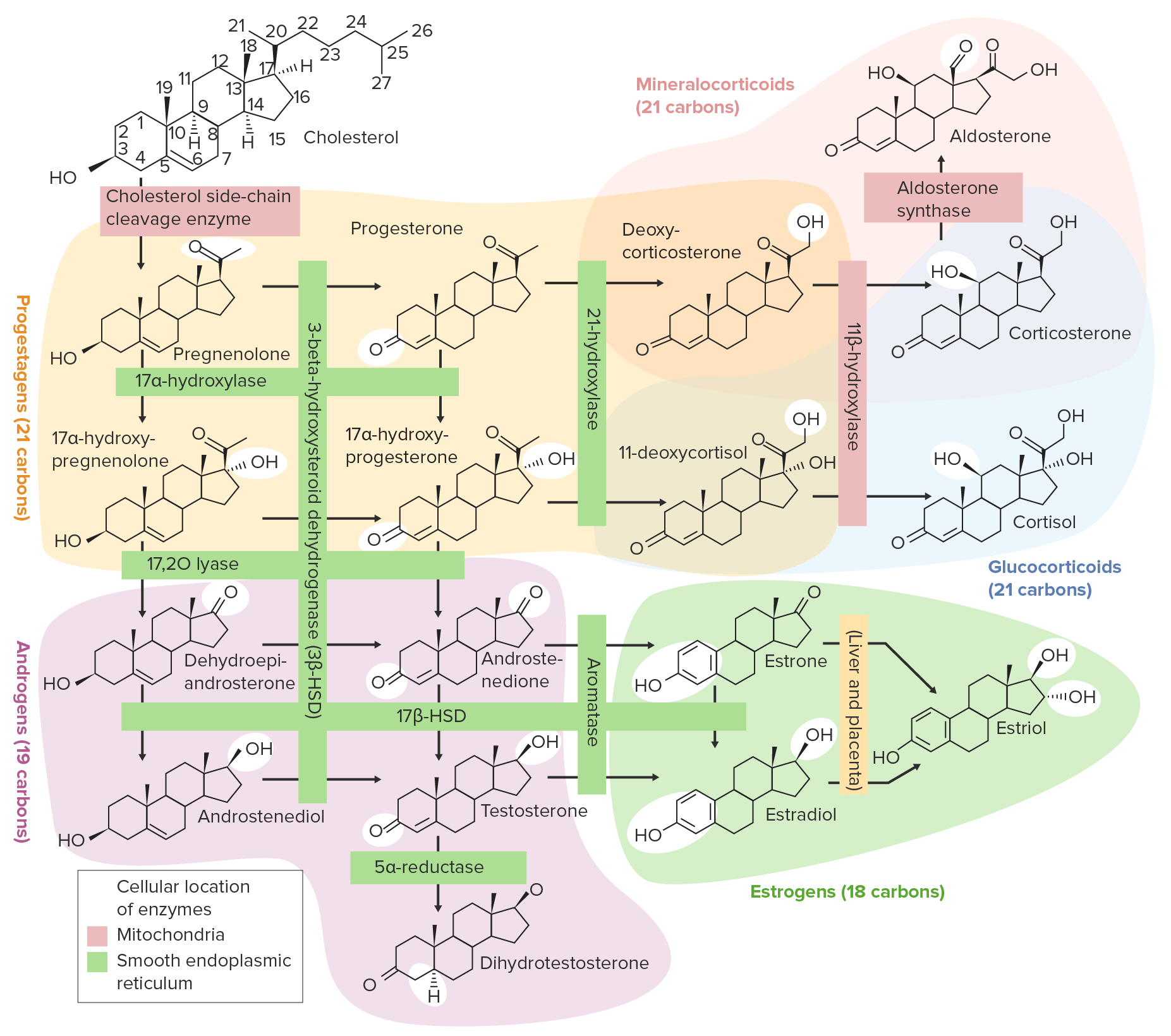
Overview of the steroidogenesis pathways:
Cholesterol is initially converted to pregnenolone and then to progesterone. From there, the pathway diverges, with many potential end products, including glucocorticoids, mineralocorticoids, androgens, and estrogens.
Image by Lecturio.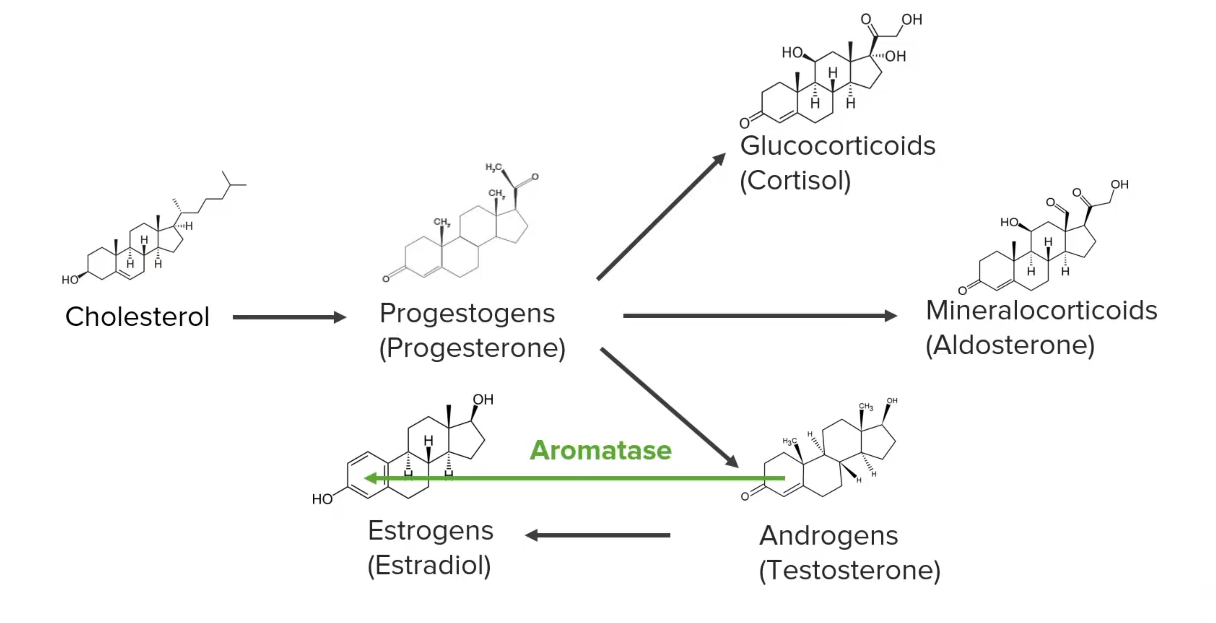
Diagram showing a simplified version of the synthesis pathways of steroid hormones
Image by Lecturio.|
Synthesis
Synthesis
Polymerase Chain Reaction (PCR) location |
Class | Primary hormone | Functions |
|---|---|---|---|
| Adrenal glands Adrenal Glands The adrenal glands are a pair of retroperitoneal endocrine glands located above the kidneys. The outer parenchyma is called the adrenal cortex and has 3 distinct zones, each with its own secretory products. Beneath the cortex lies the adrenal medulla, which secretes catecholamines involved in the fight-or-flight response. Adrenal Glands: Anatomy | Glucocorticoids Glucocorticoids Glucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs. Glucocorticoids | Cortisone Cortisone A naturally occurring glucocorticoid that has been used in replacement therapy for adrenal insufficiency and as an anti-inflammatory agent. Cortisone itself is inactive; it is converted in the liver to the active metabolite hydrocortisone. Glucocorticoids |
|
| Mineralocorticoids Mineralocorticoids Mineralocorticoids are a drug class within the corticosteroid family and fludrocortisone is the primary medication within this class. Fludrocortisone is a fluorinated analog of cortisone. The fluorine moiety protects the drug from isoenzyme inactivation in the kidney, allowing it to exert its mineralocorticoid effect. Mineralocorticoids | Aldosterone Aldosterone A hormone secreted by the adrenal cortex that regulates electrolyte and water balance by increasing the renal retention of sodium and the excretion of potassium. Hyperkalemia |
|
|
| Gonads Gonads The gamete-producing glands, ovary or testis. Hormones: Overview and Types | Progestins Progestins Compounds that interact with progesterone receptors in target tissues to bring about the effects similar to those of progesterone. Primary actions of progestins, including natural and synthetic steroids, are on the uterus and the mammary gland in preparation for and in maintenance of pregnancy. Hormonal Contraceptives | Progesterone Progesterone The major progestational steroid that is secreted primarily by the corpus luteum and the placenta. Progesterone acts on the uterus, the mammary glands and the brain. It is required in embryo implantation; pregnancy maintenance, and the development of mammary tissue for milk production. Progesterone, converted from pregnenolone, also serves as an intermediate in the biosynthesis of gonadal steroid hormones and adrenal corticosteroids. Gonadal Hormones |
|
| Estrogens | Estradiol Estradiol The 17-beta-isomer of estradiol, an aromatized C18 steroid with hydroxyl group at 3-beta- and 17-beta-position. Estradiol-17-beta is the most potent form of mammalian estrogenic steroids. Noncontraceptive Estrogen and Progestins | Sexual development, differentiation, and maturation of primary and secondary sex characteristics Secondary sex characteristics Gonadal Hormones | |
| Androgens Androgens Androgens are naturally occurring steroid hormones responsible for development and maintenance of the male sex characteristics, including penile, scrotal, and clitoral growth, development of sexual hair, deepening of the voice, and musculoskeletal growth. Androgens and Antiandrogens | Testosterone Testosterone A potent androgenic steroid and major product secreted by the leydig cells of the testis. Its production is stimulated by luteinizing hormone from the pituitary gland. In turn, testosterone exerts feedback control of the pituitary LH and FSH secretion. Depending on the tissues, testosterone can be further converted to dihydrotestosterone or estradiol. Androgens and Antiandrogens |