Alpha-1 antitrypsin deficiency is a genetic disorder caused by inherited genetic mutations Genetic Mutations Carcinogenesis of the SERPINA1 gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics, causing the defective production of the protease Protease Enzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene. HIV Infection and AIDS inhibitor alpha-1 antitrypsin (AAT). These mutations can lead to deficiency of the enzyme causing lung disease, production of an abnormal form of the enzyme leading to liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy dysfunction, or both. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship commonly present with emphysema Emphysema Enlargement of air spaces distal to the terminal bronchioles where gas-exchange normally takes place. This is usually due to destruction of the alveolar wall. Pulmonary emphysema can be classified by the location and distribution of the lesions. Chronic Obstructive Pulmonary Disease (COPD), spontaneous pneumothorax Pneumothorax A pneumothorax is a life-threatening condition in which air collects in the pleural space, causing partial or full collapse of the lung. A pneumothorax can be traumatic or spontaneous. Patients present with a sudden onset of sharp chest pain, dyspnea, and diminished breath sounds on exam. Pneumothorax, cirrhosis Cirrhosis Cirrhosis is a late stage of hepatic parenchymal necrosis and scarring (fibrosis) most commonly due to hepatitis C infection and alcoholic liver disease. Patients may present with jaundice, ascites, and hepatosplenomegaly. Cirrhosis can also cause complications such as hepatic encephalopathy, portal hypertension, portal vein thrombosis, and hepatorenal syndrome. Cirrhosis, hepatitis, or hepatocellular carcinoma Hepatocellular carcinoma Hepatocellular carcinoma (HCC) typically arises in a chronically diseased or cirrhotic liver and is the most common primary liver cancer. Diagnosis may include ultrasound, CT, MRI, biopsy (if inconclusive imaging), and/or biomarkers. Hepatocellular Carcinoma (HCC) and Liver Metastases. There is no known cure. Management is supportive and includes infusion of AAT, treatment of comorbidities Comorbidities The presence of co-existing or additional diseases with reference to an initial diagnosis or with reference to the index condition that is the subject of study. Comorbidity may affect the ability of affected individuals to function and also their survival; it may be used as a prognostic indicator for length of hospital stay, cost factors, and outcome or survival. St. Louis Encephalitis Virus, and liver transplantation Liver transplantation The transference of a part of or an entire liver from one human or animal to another. Hepatocellular Carcinoma (HCC) and Liver Metastases. Prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas may vary based on the form of disease contracted and the severity of symptoms.
Last updated: Apr 25, 2025
Liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy disease:
Lung disease:
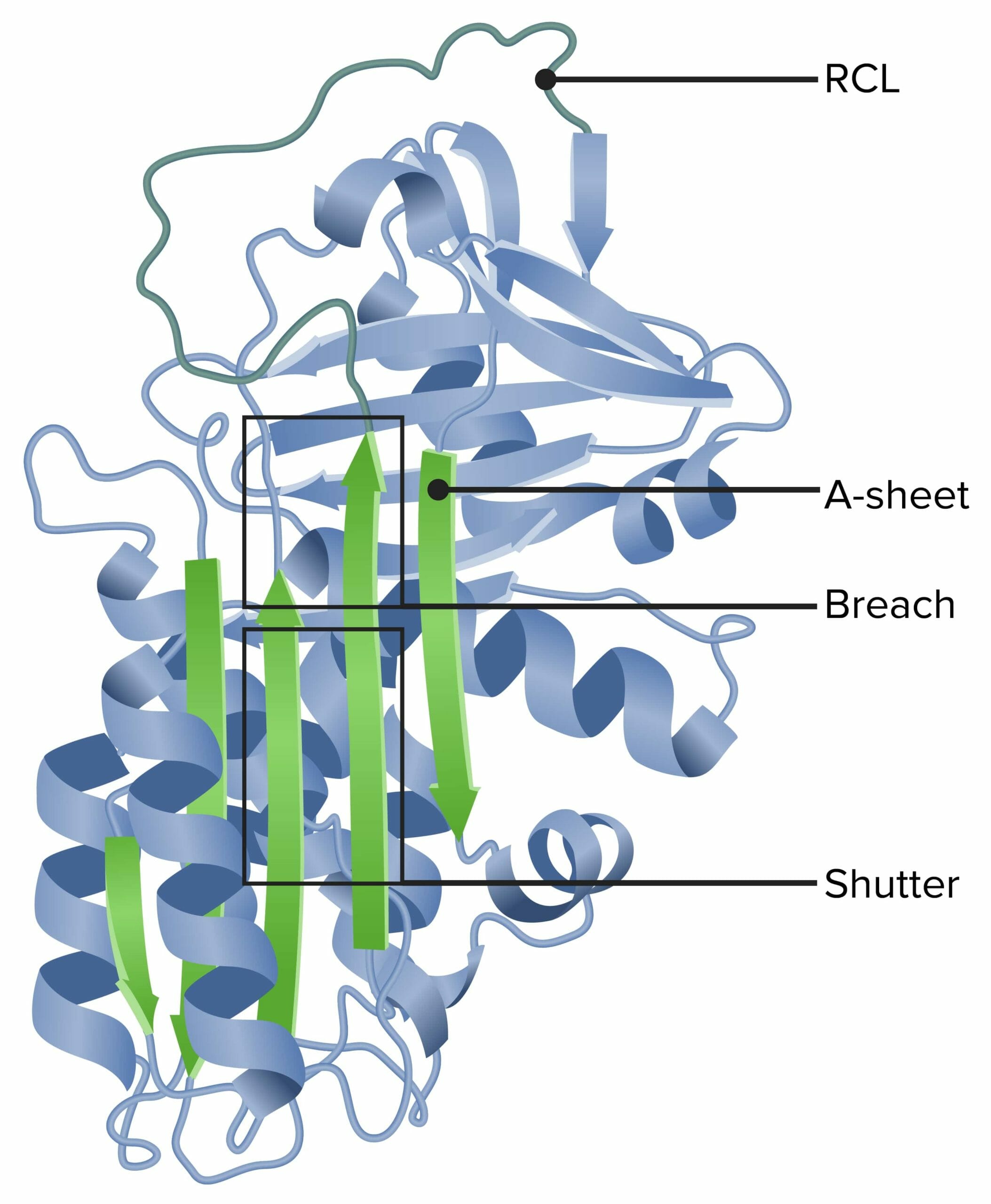
Alpha-1 antitrypsin
RCL: reactive center loop
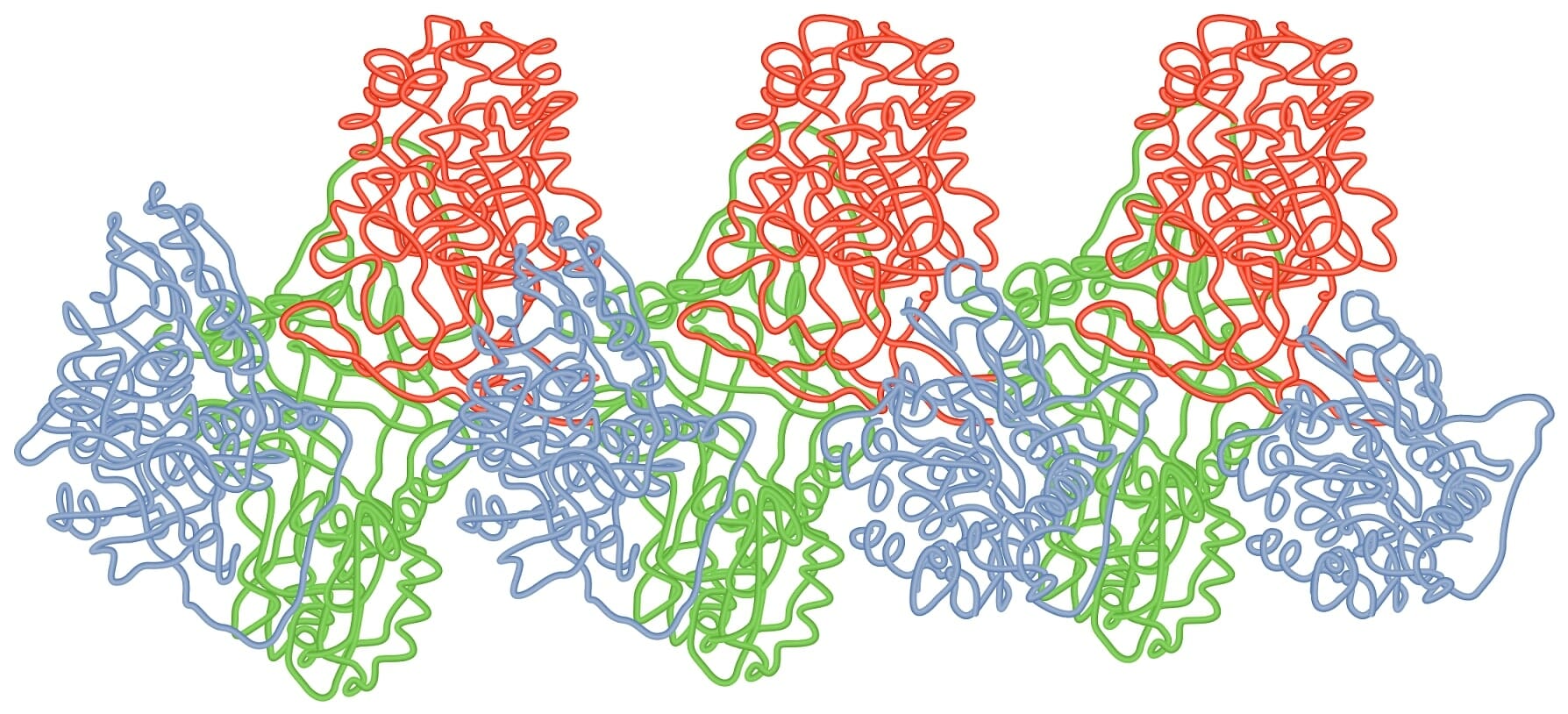
Loop-sheet polymer of AAT molecules
Image by Lecturio.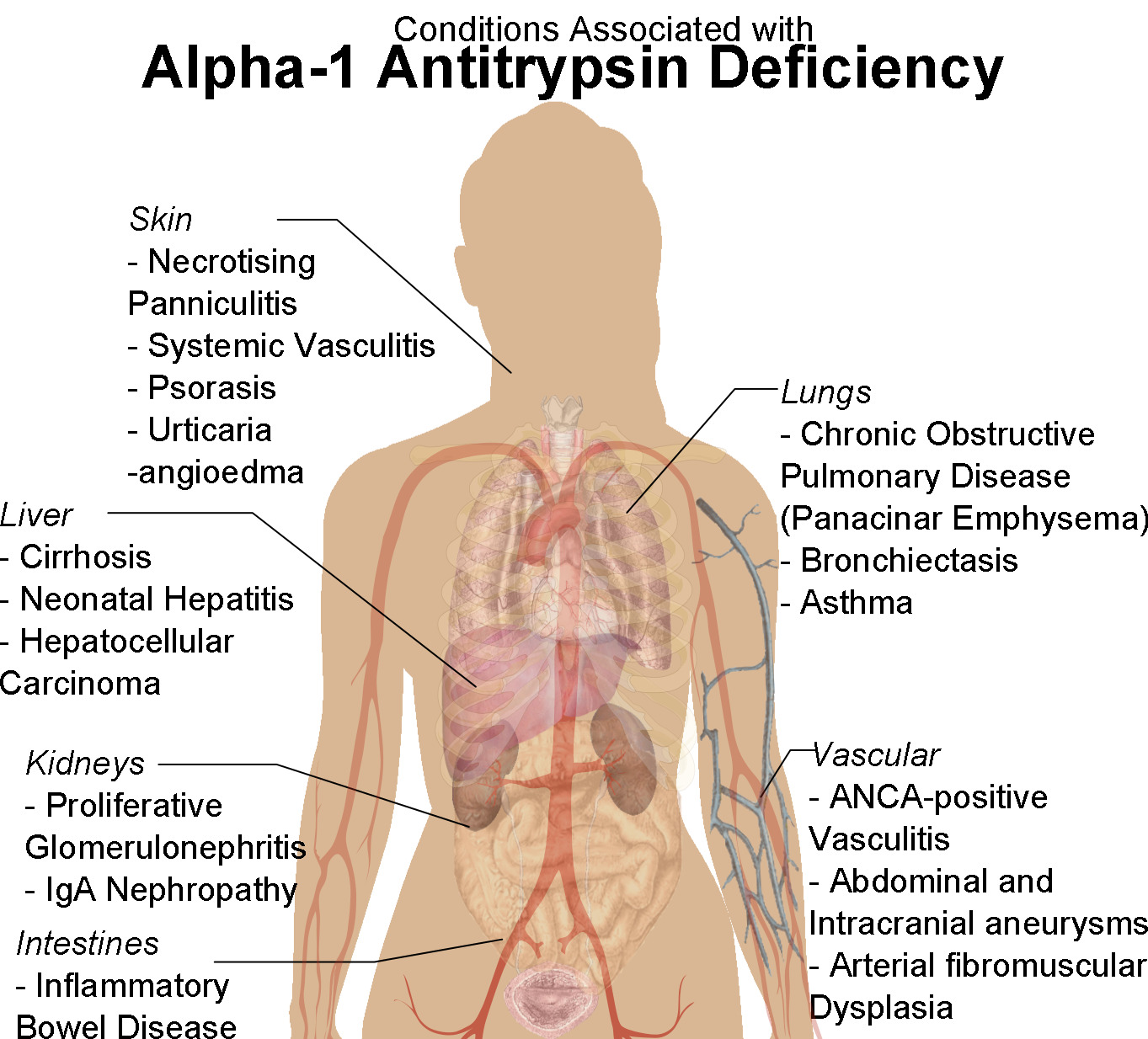
Human female shadow diagram with conditions associated with alpha-1 antitrypsin deficiency
Image: “Human female shadow diagram with conditions associated with Alpha-1 Antitrypsin Deficiency” by Mikael Häggström. License: CC0 1.0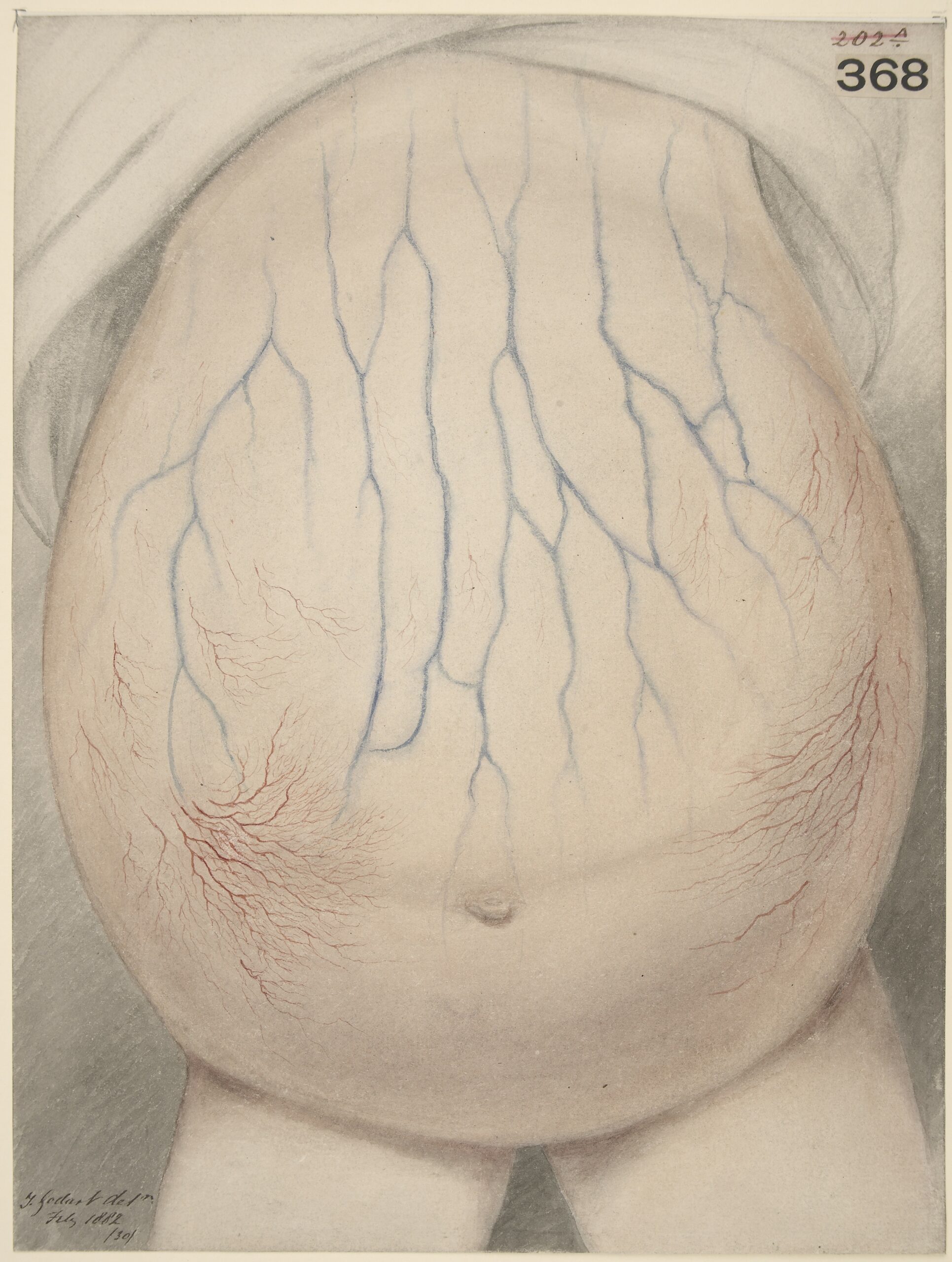
Abdomen of child affected with ascites
Image: “Watercolour drawing of the abdomen of a child affected with ascites” by Wellcome Collection gallery. License: CC BY 4.0