Cystic Cystic Fibrocystic Change fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans is an autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance disorder caused by mutations in the gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics CFTR. The mutations lead to dysfunction of chloride Chloride Inorganic compounds derived from hydrochloric acid that contain the Cl- ion. Electrolytes channels Channels The Cell: Cell Membrane, which results in hyperviscous mucus and the accumulation of secretions. Common presentations include chronic respiratory infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease, failure to thrive Failure to Thrive Failure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive, and pancreatic insufficiency. The gold standard for diagnosis is the sweat chloride Chloride Inorganic compounds derived from hydrochloric acid that contain the Cl- ion. Electrolytes test, which can be complemented by genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies. Cystic Cystic Fibrocystic Change fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans ultimately leads to chronic inflammation Chronic Inflammation Inflammation and multisystem organ failure. Management includes CFTR modulator therapy and system-specific strategies for supportive care. Prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas varies depending on treatment and complications. With optimal medical care Medical care Conflict of Interest, patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship can live into their mid-40s.
Last updated: Dec 15, 2025

Types of mutation in cystic fibrosis
Image by Lecturio.Impact on organ systems
| Small intestine Small intestine The small intestine is the longest part of the GI tract, extending from the pyloric orifice of the stomach to the ileocecal junction. The small intestine is the major organ responsible for chemical digestion and absorption of nutrients. It is divided into 3 segments: the duodenum, the jejunum, and the ileum. Small Intestine: Anatomy | Thick secretions impair absorption Absorption Absorption involves the uptake of nutrient molecules and their transfer from the lumen of the GI tract across the enterocytes and into the interstitial space, where they can be taken up in the venous or lymphatic circulation. Digestion and Absorption, increasing the risk of obstruction. |
|
| Large intestine Large intestine The large intestines constitute the last portion of the digestive system. The large intestine consists of the cecum, appendix, colon (with ascending, transverse, descending, and sigmoid segments), rectum, and anal canal. The primary function of the colon is to remove water and compact the stool prior to expulsion from the body via the rectum and anal canal. Colon, Cecum, and Appendix: Anatomy | Incompletely digested macronutrients lead to thick stool, predisposing patient to impaction, obstruction, intussusception Intussusception Intussusception occurs when a part of the intestine (intussusceptum) telescopes into another part (intussuscipiens) of the intestine. The condition can cause obstruction and, if untreated, progress to bowel ischemia. Intussusception is most common in the pediatric population, but is occasionally encountered in adults. Intussusception. | |
| Pancreas Pancreas The pancreas lies mostly posterior to the stomach and extends across the posterior abdominal wall from the duodenum on the right to the spleen on the left. This organ has both exocrine and endocrine tissue. Pancreas: Anatomy |
|
Abdominal pain Abdominal Pain Acute Abdomen; abdominal cramping; bloating Bloating Constipation; frequent bulky, oily stools; weight loss Weight loss Decrease in existing body weight. Bariatric Surgery; flatulence |
| Hepatobiliary |
|
Right upper quadrant Right upper quadrant Anterior Abdominal Wall: Anatomy pain Pain An unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons. Pain: Types and Pathways after large, fatty meals; nausea Nausea An unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses. Antiemetics; vomiting Vomiting The forcible expulsion of the contents of the stomach through the mouth. Hypokalemia; jaundice Jaundice Jaundice is the abnormal yellowing of the skin and/or sclera caused by the accumulation of bilirubin. Hyperbilirubinemia is caused by either an increase in bilirubin production or a decrease in the hepatic uptake, conjugation, or excretion of bilirubin. Jaundice |
To recall the most common clinical features of cystic Cystic Fibrocystic Change fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans, remember the acronym CF PANCREAS Pancreas The pancreas lies mostly posterior to the stomach and extends across the posterior abdominal wall from the duodenum on the right to the spleen on the left. This organ has both exocrine and endocrine tissue. Pancreas: Anatomy:
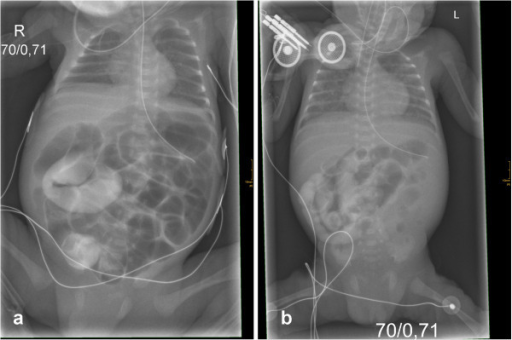
A lethal course of meconium ileus in preterm twins revealing a novel cystic fibrosis mutation
Image: “Initial X-rays of both twins” by Department of Pediatrics, Albert-Ludwigs-University of Freiburg, Mathildenstrasse 1, D 79106 Freiburg, Germany. alexander.puzik@uniklinik-freiburg.de. License: CC BY 2.0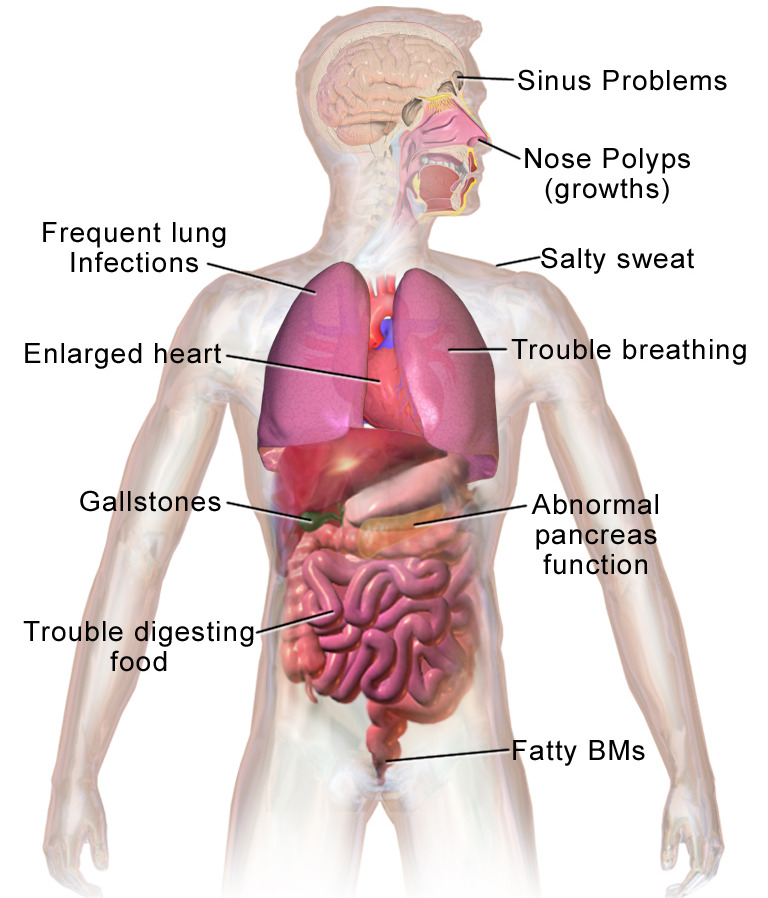
Most common clinical presentations of cystic fibrosis
BMs: bowel movements
| Exacerbation of pulmonary disease Pulmonary disease Diseases involving the respiratory system. Blastomyces/Blastomycosis |
|
|---|---|
| Chronic microbial colonization Colonization Bacteriology |
|
| Allergic bronchopulmonary aspergillosis Allergic bronchopulmonary aspergillosis Hypersensitivity reaction (allergic reaction) to fungus aspergillus in an individual with long-standing bronchial asthma. It is characterized by pulmonary infiltrates, eosinophilia, elevated serum immunoglobulin e, and skin reactivity to aspergillus antigen. Aspergillus/Aspergillosis | Aspergillus Aspergillus A genus of mitosporic fungi containing about 100 species and eleven different teleomorphs in the family trichocomaceae. Echinocandins airway Airway ABCDE Assessment colonization Colonization Bacteriology, followed by vigorous IgE- and IgG-mediated immune response, varied presentation which can include worsening fever Fever Fever is defined as a measured body temperature of at least 38°C (100.4°F). Fever is caused by circulating endogenous and/or exogenous pyrogens that increase levels of prostaglandin E2 in the hypothalamus. Fever is commonly associated with chills, rigors, sweating, and flushing of the skin. Fever, malaise Malaise Tick-borne Encephalitis Virus, mucous plugging that may not improve with antibiotics |
| Biliary cirrhosis Cirrhosis Cirrhosis is a late stage of hepatic parenchymal necrosis and scarring (fibrosis) most commonly due to hepatitis C infection and alcoholic liver disease. Patients may present with jaundice, ascites, and hepatosplenomegaly. Cirrhosis can also cause complications such as hepatic encephalopathy, portal hypertension, portal vein thrombosis, and hepatorenal syndrome. Cirrhosis | Bile Bile An emulsifying agent produced in the liver and secreted into the duodenum. Its composition includes bile acids and salts; cholesterol; and electrolytes. It aids digestion of fats in the duodenum. Gallbladder and Biliary Tract: Anatomy duct obstruction in adults |
| Endocrine pancreatic insufficiency | Development of diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus mellitus, including the destruction of the islets of Langerhans in adulthood |
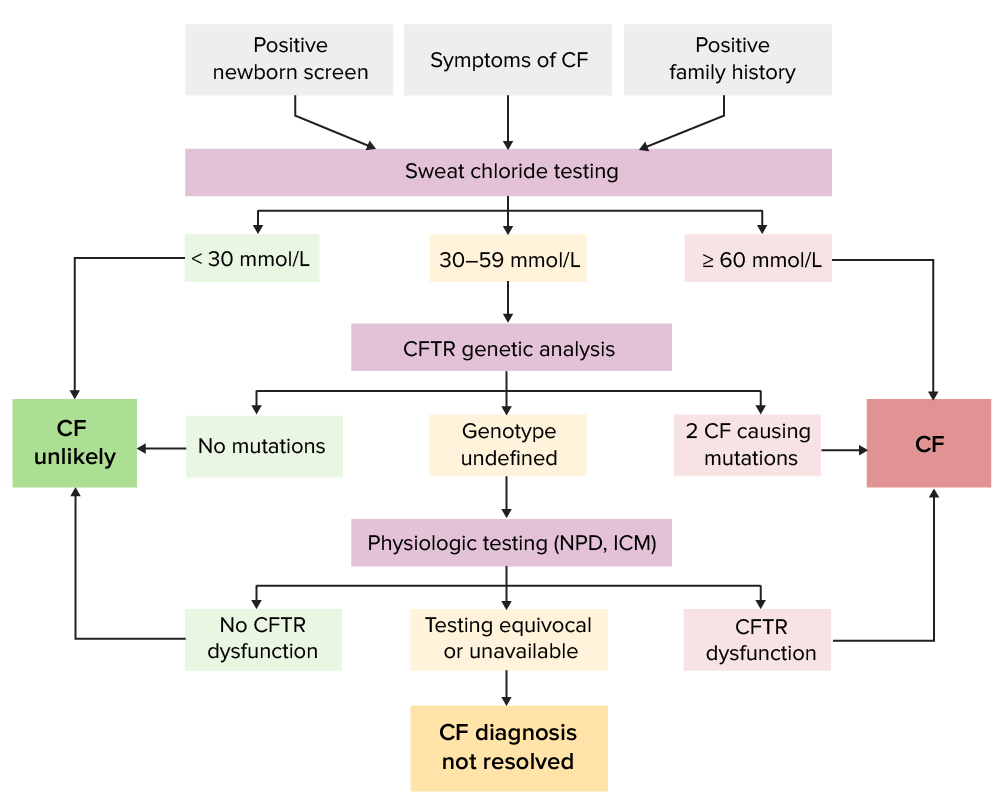
Algorithm for cystic fibrosis diagnosis
Image by Lecturio.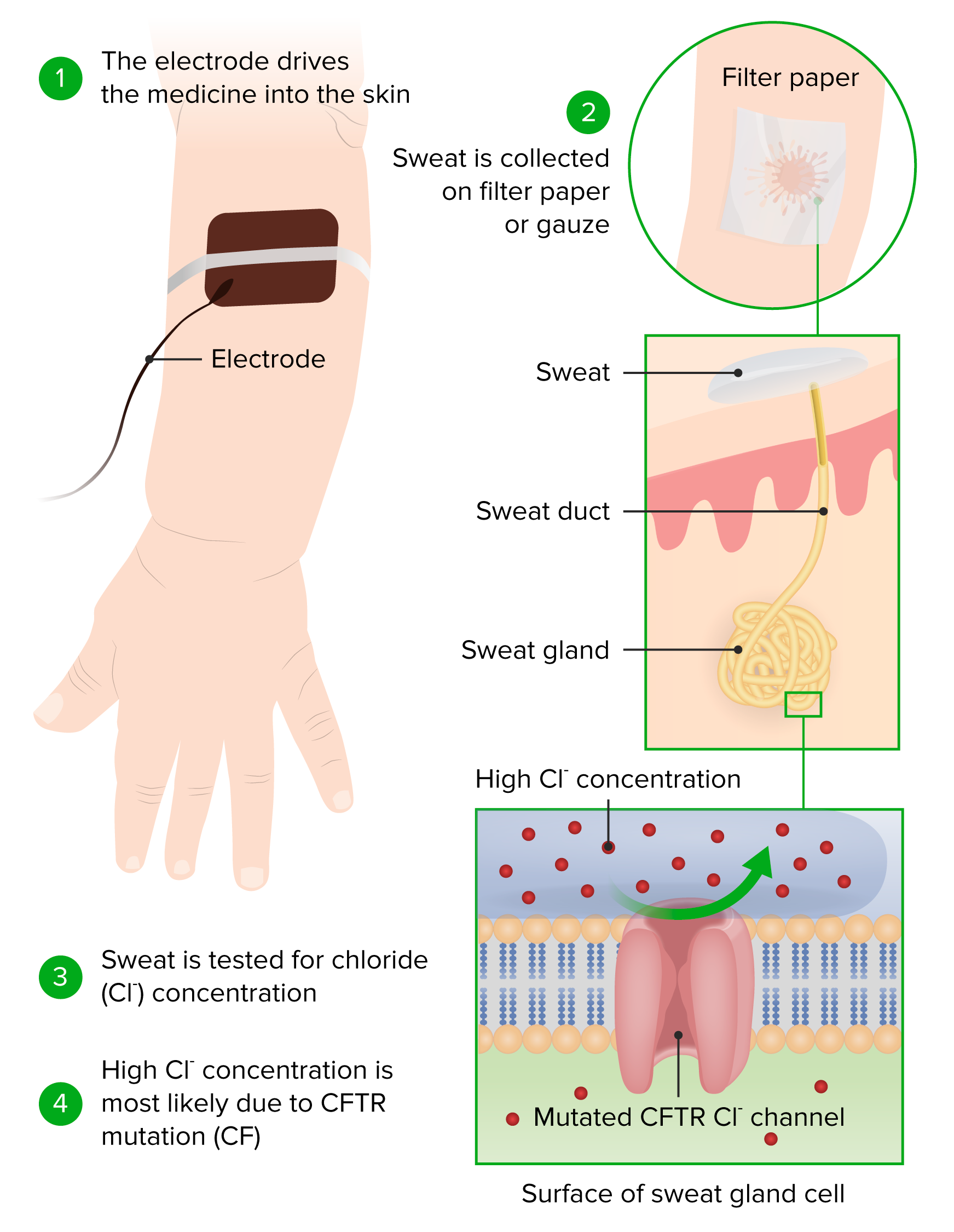
Principle and conduction of a sweat chloride test
Image by Lecturio.If the diagnosis remains unclear, further testing can be done.
Nasal potential difference:
Intestinal current measurements (ICM):
Fecal elastase Elastase A protease of broad specificity, obtained from dried pancreas. Molecular weight is approximately 25, 000. The enzyme breaks down elastin, the specific protein of elastic fibers, and digests other proteins such as fibrin, hemoglobin, and albumin. Proteins and Peptides:
Pancreatic insufficiency can be detected when screening Screening Preoperative Care the stool for pancreatic elastase-1 Elastase-1 A protease of broad specificity, obtained from dried pancreas. Molecular weight is approximately 25, 000. The enzyme breaks down elastin, the specific protein of elastic fibers, and digests other proteins such as fibrin, hemoglobin, and albumin. Pancreatic Parameters, which is absent in 80% of people with CF.
Imaging:
Airway Airway ABCDE Assessment culture:
Management of CF should be multidisciplinary and include specialized physicians Physicians Individuals licensed to practice medicine. Clinician–Patient Relationship, physiotherapists, dieticians, and/or psychological support.
| Organ system | Approach to therapy |
|---|---|
| Lung infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease, respiratory lung insufficiency |
|
| Hypotonic Hypotonic Solutions that have a lesser osmotic pressure than a reference solution such as blood, plasma, or interstitial fluid. Renal Sodium and Water Regulation dehydration Dehydration The condition that results from excessive loss of water from a living organism. Volume Depletion and Dehydration/hypochloremic alkalosis Alkalosis A pathological condition that removes acid or adds base to the body fluids. Respiratory Alkalosis |
|
| Exocrine pancreatic insufficiency Exocrine pancreatic insufficiency A malabsorption condition resulting from greater than 10% reduction in the secretion of pancreatic digestive enzymes (lipase; proteases; and amylase) by the exocrine pancreas into the duodenum. This condition is often associated with cystic fibrosis and with chronic pancreatitis. Malabsorption and Maldigestion | A dose of pancreatic enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes (defined lipase Lipase An enzyme of the hydrolase class that catalyzes the reaction of triacylglycerol and water to yield diacylglycerol and a fatty acid anion. It is produced by glands on the tongue and by the pancreas and initiates the digestion of dietary fats. Malabsorption and Maldigestion and protease Protease Enzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene. HIV Infection and AIDS concentration) with every meal |
| Failure to thrive Failure to Thrive Failure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive |
|
| Vitamin and mineral deficits | Prophylactic substitution of liposoluble vitamins in supranormal doses, replace minerals Minerals Electrolytes and trace elements Trace elements Trace elements are minerals required in small amounts (1-100 mg/day in adults) to carry out biologic functions. These elements act as cofactors for essential enzymes as well as being components of hormones and antioxidant molecules. Iron, chromium, copper, and iodine are among these elements. Trace Elements |
| CF-related diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus |
|
The following conditions are related to cystic Cystic Fibrocystic Change fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans: