La fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans quística es un trastorno autosómico recesivo causado por mutaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen regulador de conductancia transmembrana de la fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans quística (CFTR, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés). Las mutaciones conducen a una disfunción de los LOS Neisseria canales de cloruro, lo que da lugar a una mucosidad hiperviscosa y acumulación de secreciones. Las presentaciones más comunes incluyen; las infecciones respiratorias crónicas, retraso en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el desarrollo e insuficiencia pancreática. El estándar de oro para el diagnóstico es la prueba de cloruro en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el sudor, que puede complementarse con pruebas genéticas. La fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans quística acaba provocando una inflamación crónica y un fallo orgánico multisistémico. El tratamiento incluye la terapia con moduladores de CFTR y estrategias específicas de sistemas para el tratamiento de soporte. El pronóstico varía según el tratamiento y las complicaciones. Con una atención médica óptima, los LOS Neisseria pacientes pueden vivir hasta mediados de los LOS Neisseria 40 años.
Last updated: Dec 15, 2025

Tipos de mutaciones en la fibrosis quística
Imagen por Lecturio.Impacto en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria sistemas de órganos
| Intestino delgado | Las secreciones espesas dificultan la absorción, aumentando el riesgo de obstrucción. |
|
| Intestino grueso | Los LOS Neisseria macronutrientes mal digeridos dan lugar a heces espesas, lo que predispone al AL Amyloidosis paciente a la impactación, obstrucción e intususcepción. | |
| Páncreas |
|
Dolor Dolor Inflammation abdominal; calambres abdominales; hinchazón; heces frecuentes voluminosas y aceitosas; pérdida de peso; flatulencia |
| Hepatobiliar |
|
Dolor Dolor Inflammation en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el cuadrante superior derecho después de comidas copiosas y grasas; náuseas; vómitos; ictericia |
Para recordar las características clínicas más comunes de la fibrosis quística, recuerde el acrónimo “CF PANCREAS” ( en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés):
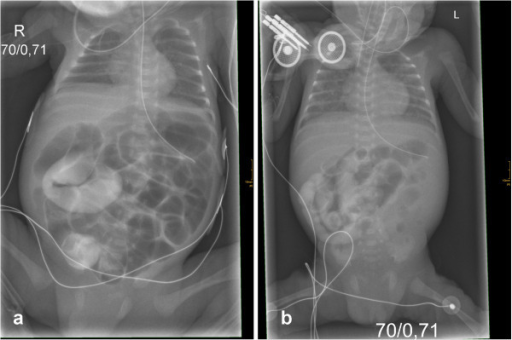
Una evolución letal del íleo meconial en gemelos prematuros que revela una nueva mutación de la fibrosis quística
Imagen: “Initial X-rays of both twins” por Department of Pediatrics, Albert-Ludwigs-University of Freiburg, Mathildenstrasse 1, D 79106 Freiburg, Germany. alexander.puzik@uniklinik-freiburg.de.Licencia: CC BY 2.0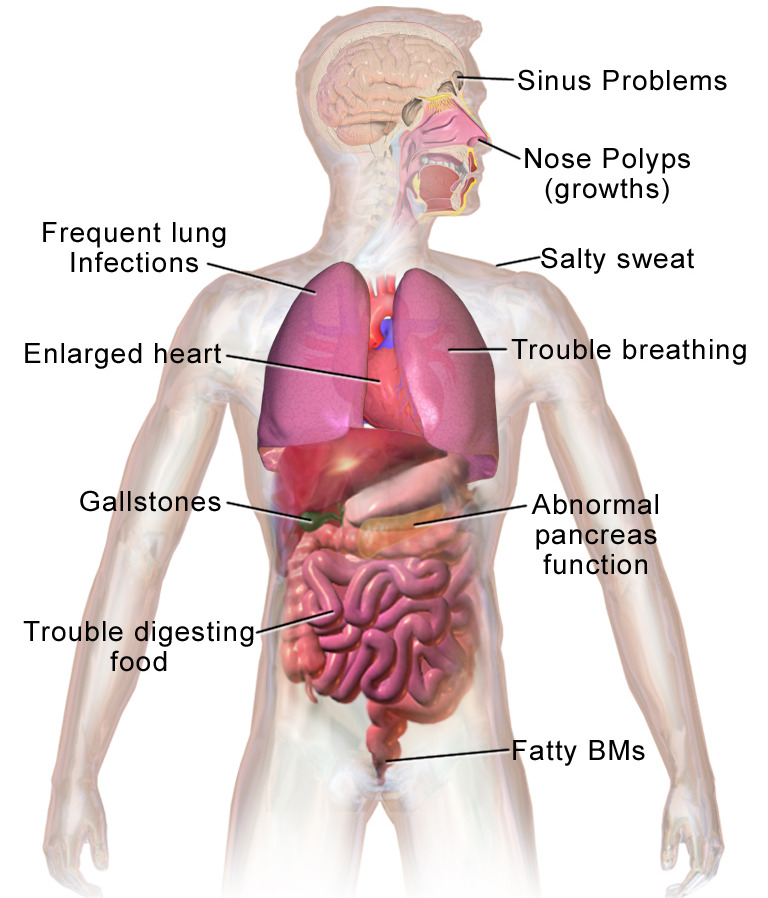
Presentaciones clínicas más comunes de la fibrosis quística
Movimientos intestinales
| Exacerbación de la enfermedad pulmonar |
|
|---|---|
| Colonización microbiana crónica |
|
| Aspergilosis broncopulmonar alérgica | Colonización de las vías respiratorias por Aspergillus Aspergillus A genus of mitosporic fungi containing about 100 species and eleven different teleomorphs in the family trichocomaceae. Echinocandins, seguida de una vigorosa respuesta inmunitaria mediada por IgE IgE An immunoglobulin associated with mast cells. Overexpression has been associated with allergic hypersensitivity. Immunoglobulins: Types and Functions e IgG IgG The major immunoglobulin isotype class in normal human serum. There are several isotype subclasses of igg, for example, igg1, igg2a, and igg2b. Hypersensitivity Pneumonitis, presentación variada que puede incluir empeoramiento de la fiebre, malestar, taponamiento de la mucosa que puede no mejorar con antibióticos |
| Cirrosis biliar | Obstrucción de la vía biliar en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum adultos |
| Insuficiencia pancreática endocrina | Desarrollo de diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus mellitus, incluida la destrucción de los LOS Neisseria islotes de Langerhans en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la adultez |
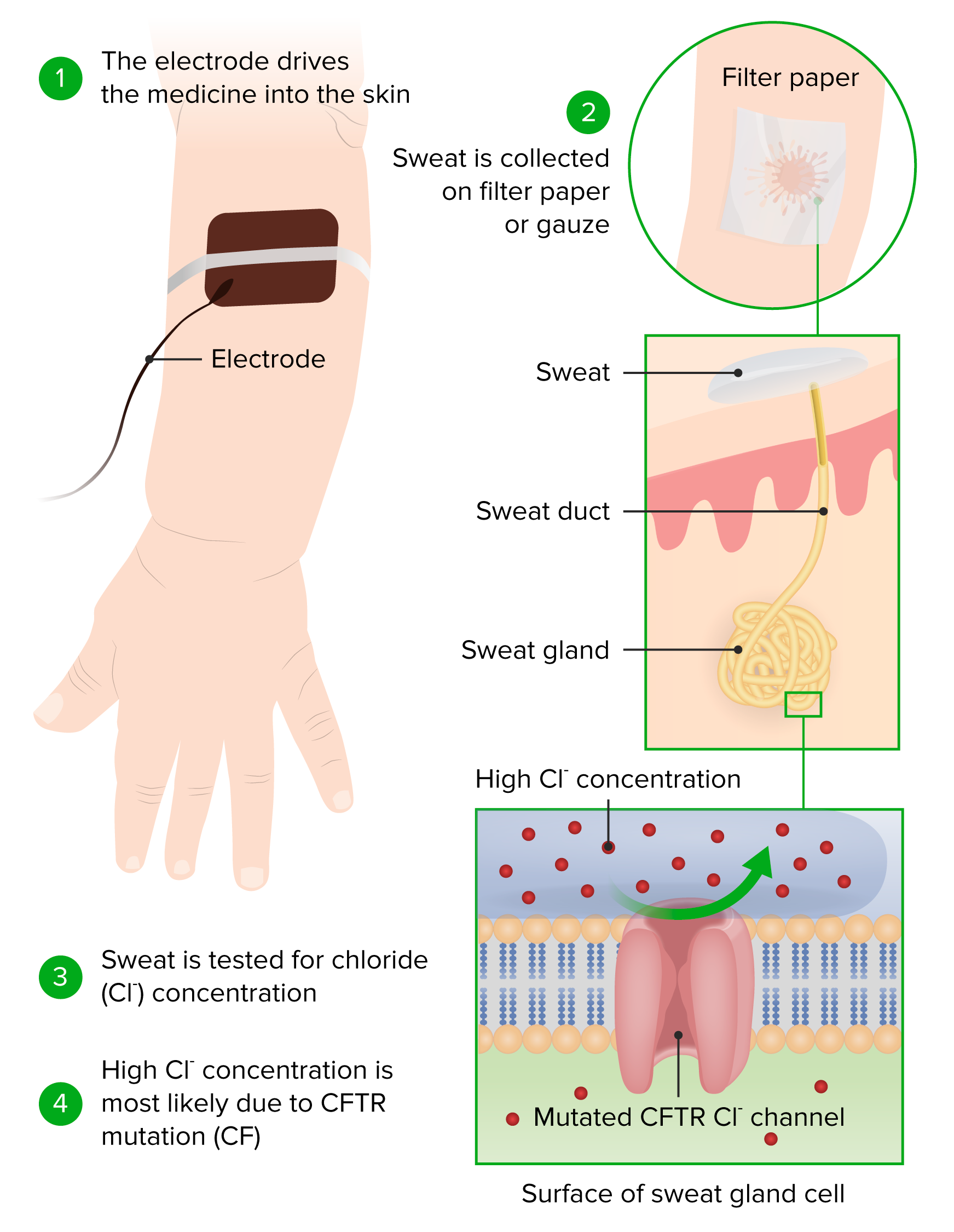
Principios de la prueba de cloruro en el sudor y su realización
Imagen por Lecturio.Si el diagnóstico sigue sin estar claro, se pueden realizar más pruebas.
Diferencia de potencial nasal:
Mediciones de corriente intestinal:
Elastasa fecal:
La insuficiencia pancreática puede detectarse al AL Amyloidosis examinar las heces en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum busca de elastasa-1 pancreática, que está ausente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el 80% de las personas con FQ.
Imagenología:
Cultivo de las vías respiratorias:
El tratamiento de la FQ debe ser multidisciplinar e incluir médicos especialistas, fisioterapeutas, dietistas y/o apoyo psicológico.
| Sistema de órganos | Abordaje de la terapia |
|---|---|
| Infecciones pulmonares, insuficiencia pulmonar respiratoria |
|
| Deshidratación hipotónica/alcalosis hipoclorémica |
|
| Insuficiencia pancreática exocrina | Una dosis de enzimas pancreáticas (concentración definida de lipasa y proteasa) con cada comida |
| Retraso en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el desarrollo |
|
| Déficit de vitaminas y minerales | Sustitución profiláctica de vitaminas liposolubles en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum dosis supranormales, sustitución de minerales y oligoelementos |
| Diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus relacionada con la FQ |
|
Las siguientes afecciones están relacionadas con la fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans quística: