


A fibrose quística é um distúrbio autossómico recessivo causado por mutações no gene CFTR. As mutações levam à disfunção dos canais de cloro, resultando num muco hiperviscoso e na acumulação de secreções. As apresentações clínicas mais comuns incluem infeções respiratórias crónicas, má evolução estaturo-ponderal e insuficiência pancreática. O exame de diagnóstico "gold standard" é o teste de cloro no suor, e pode ser complementado por testes Testes Gonadal Hormones genéticos. A fibrose quística leva à inflamação crónica e falência multiorgânica. O tratamento inclui terapêutica do modulador do CFTR e tratamento de suporte dirigido aos órgãos afetados. O prognóstico varia conforme o tratamento e complicações. Com um ótimo acompanhamento médico, os pacientes podem viver até aos 40 anos.
Last updated: Dec 15, 2025

Tipos de mutação na fibrose quística
Imagem por Lecturio.Impacto nos órgãos
| Intestino delgado | Apresentam secreções espessas que prejudicam a absorção intestinal, aumentando o risco de obstrução. |
|
| Intestino grosso | Os macronutrientes incompletamente digeridos resultam em fezes espessas, predispondo o paciente a obstrução, impactação fecal e intussusceção intestinal. | |
| Pâncreas |
|
Dor abdominal; cólicas abdominais; distensão abdominal; fezes volumosas e oleosas frequentes; perda de peso; flatulências |
| Hepatobiliar |
|
Dor no quadrante superior direito após refeições grandes e gordurosas; náuseas; vómitos; icterícia |
Para lembrar as características clínicas mais comuns da fibrose quística, lembre-se da sigla “CF PANCREAS“:
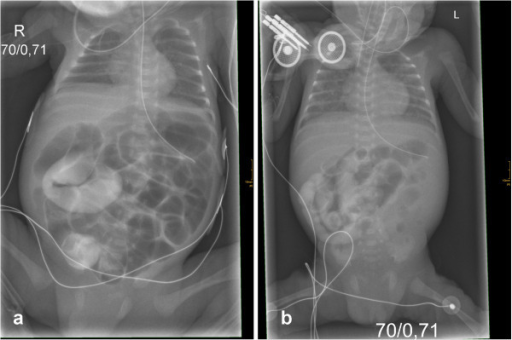
Um caso letal de ileus meconial em gémeos prematuros, apresentando uma nova mutação da fibrose quística
Imagem: “Initial X-rays of both twins” por Department of Pediatrics, Albert-Ludwigs-University of Freiburg, Mathildenstrasse 1, D 79106 Freiburg, Germany. alexander.puzik@uniklinik-freiburg.de. Licença: CC BY 2.0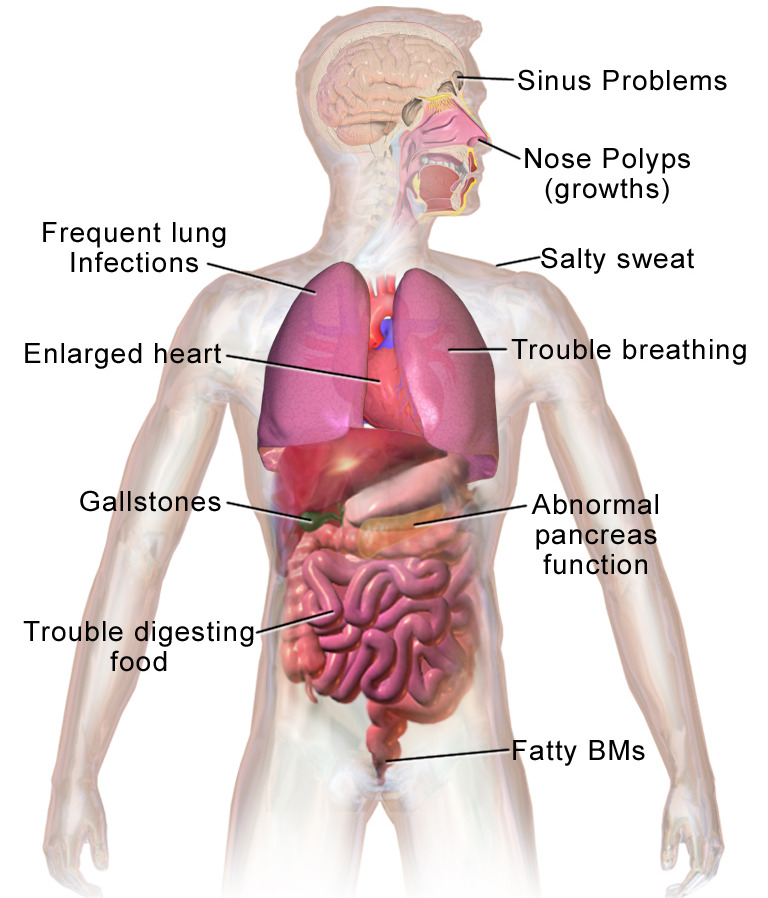
Apresentações clínicas mais comuns de fibrose quística
BMs: movimentos intestinais, pela sigla em inglês
| Exacerbação da doença pulmonar |
|
|---|---|
| Colonização crónica por microorganismos |
|
| Aspergilose broncopulmonar alérgica | Colonização das vias aéreas por Aspergillus Aspergillus A genus of mitosporic fungi containing about 100 species and eleven different teleomorphs in the family trichocomaceae. Echinocandins, seguida de uma resposta imunológica vigorosa mediada por IgE IgE An immunoglobulin associated with mast cells. Overexpression has been associated with allergic hypersensitivity. Immunoglobulins: Types and Functions e IgG IgG The major immunoglobulin isotype class in normal human serum. There are several isotype subclasses of igg, for example, igg1, igg2a, and igg2b. Hypersensitivity Pneumonitis. Os pacientes podem apresentar vários sintomas, incluindo agravamento da febre, mal-estar e obstrução mucosa que pode não melhorar com antibioterapia |
| Cirrose biliar | Obstrução do ducto biliar em adultos |
| Insuficiência pancreática endócrina | Desenvolvimento de diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus mellitus. Pode haver destruição das ilhotas de Langerhans em adultos |
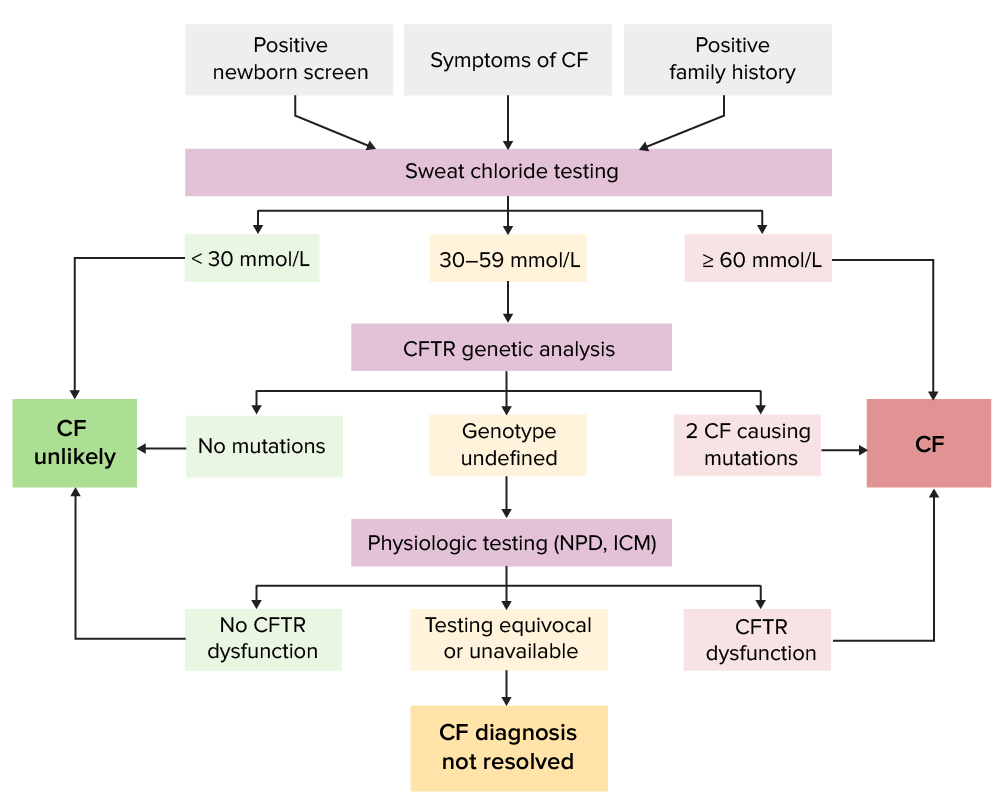
Algoritmo de diagnóstico para FQ utilizado em vários países. IRT: tripsinogénio imunoreativo, pela sigla em inglêsImagem por Lecturio.
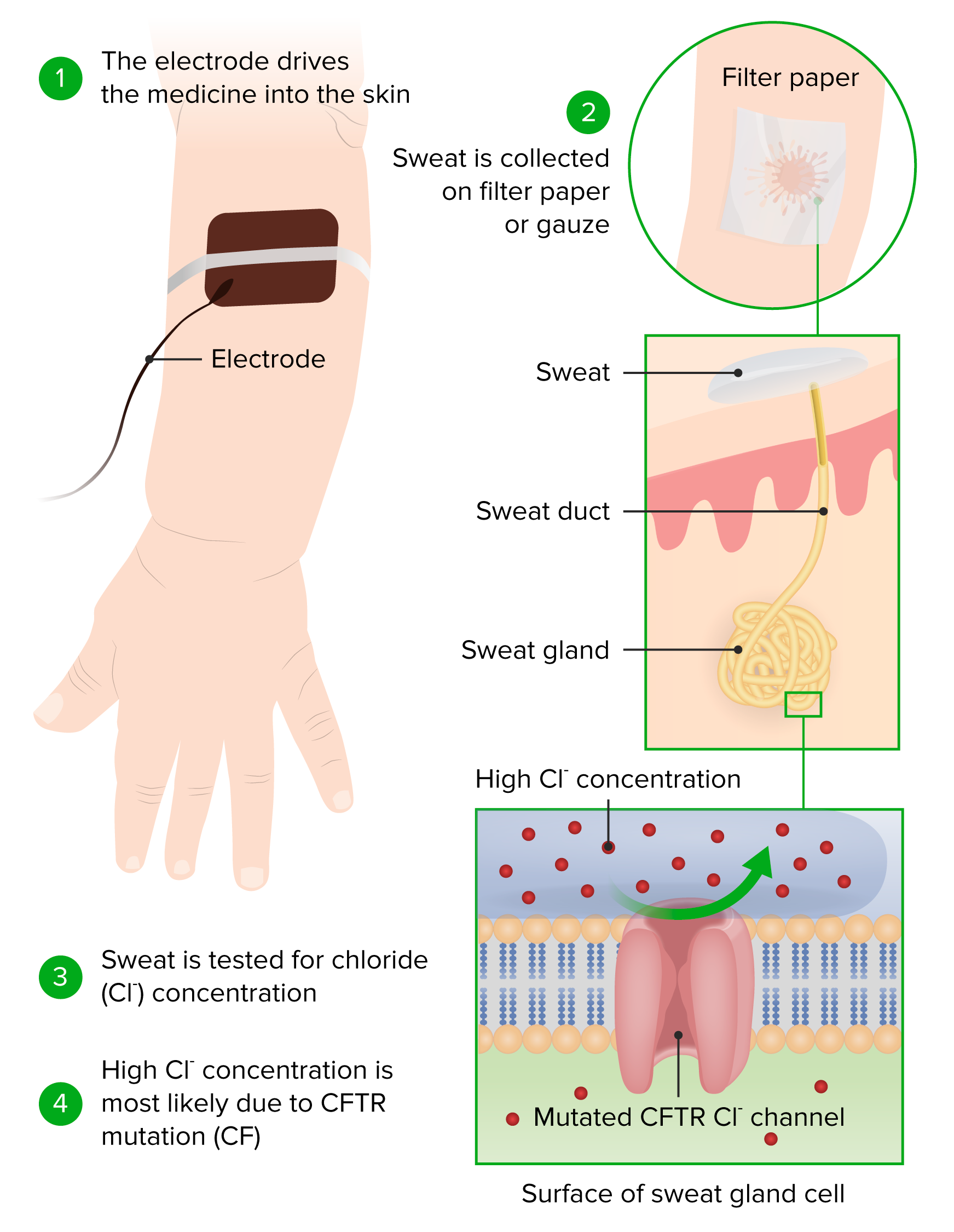
Princípio e realização de um teste de cloro de suor
Imagem por Lecturio.Se o diagnóstico não for claro, podem ser realizados mais MAIS Androgen Insensitivity Syndrome testes Testes Gonadal Hormones.
Diferença de potencial nasal:
Medição da corrente intestinal (MCI):
Elastase Elastase A protease of broad specificity, obtained from dried pancreas. Molecular weight is approximately 25, 000. The enzyme breaks down elastin, the specific protein of elastic fibers, and digests other proteins such as fibrin, hemoglobin, and albumin. Proteins and Peptides fecal:
A insuficiência pancreática pode ser detetada pela pesquisa nas fezes da elastase Elastase A protease of broad specificity, obtained from dried pancreas. Molecular weight is approximately 25, 000. The enzyme breaks down elastin, the specific protein of elastic fibers, and digests other proteins such as fibrin, hemoglobin, and albumin. Proteins and Peptides pancreática-1. No entanto, está ausente em 80% das pessoas com FQ.
Imagiologia:
Cultura de secreções das vias aéreas:
O tratamento da FQ deve ser multidisciplinar e incluir médicos especializados, fisioterapeutas, nutricionistas e/ou apoio psicológico.
| Abordagem por sistema de órgãos afetados | Abordagem terapêutica |
|---|---|
| Infeções pulmonares, insuficiência pulmonar respiratória |
|
| Desidratação hipotónica/alcalose hipoclorémica |
|
| Insuficiência pancreática exócrina | Devem ser administradas enzimas pancreáticas (concentração definida de lipase Lipase An enzyme of the hydrolase class that catalyzes the reaction of triacylglycerol and water to yield diacylglycerol and a fatty acid anion. It is produced by glands on the tongue and by the pancreas and initiates the digestion of dietary fats. Malabsorption and Maldigestion e protease Protease Enzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene. HIV Infection and AIDS) a cada refeição |
| Má progressão estaturo-ponderal |
|
| Défices vitamínicos e minerais | Suplementação profilática de vitaminas lipossolúveis em doses supranormais, suplementação com minerais e oligoelementos |
| Diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus relacionado com a FQ |
|
As seguintes condições estão relacionadas com a fibrose quística:
