Anticholinergic drugs block the effect of the neurotransmitter acetylcholine at the muscarinic receptors in the central and peripheral nervous systems. Anticholinergic agents inhibit the parasympathetic nervous system, resulting in effects on the smooth muscle in the respiratory tract, vascular system, urinary tract, GI tract, and pupils of the eyes. These medications are used in the management of a wide range of diseases, most notably in the treatment of overactive bladder, irritable bowel syndrome, chronic obstructive pulmonary disease (COPD), and allergic rhinitis. Atropine specifically is used in emergency medicine in the advanced cardiac life support (ACLS) protocol for severe bradycardia and as an antidote to organophosphate poisoning with insecticides or chemical warfare agents. Atropine is also used in anesthesiology as an antisialagogue or to reverse neuromuscular blocking agents. The term “anticholinergic” is often used to describe the adverse effects of drugs with anticholinergic properties (e.g., tricyclic antidepressants); these include dry mouth, constipation, blurred vision, and tachycardia.
Last updated: Jan 17, 2023
Atropine is the prototypical anticholinergic drug owing to its antagonism of acetylcholine Acetylcholine A neurotransmitter found at neuromuscular junctions, autonomic ganglia, parasympathetic effector junctions, a subset of sympathetic effector junctions, and at many sites in the central nervous system. Receptors and Neurotransmitters of the CNS ( ACh ACh A neurotransmitter found at neuromuscular junctions, autonomic ganglia, parasympathetic effector junctions, a subset of sympathetic effector junctions, and at many sites in the central nervous system. Receptors and Neurotransmitters of the CNS) receptors Receptors Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors.
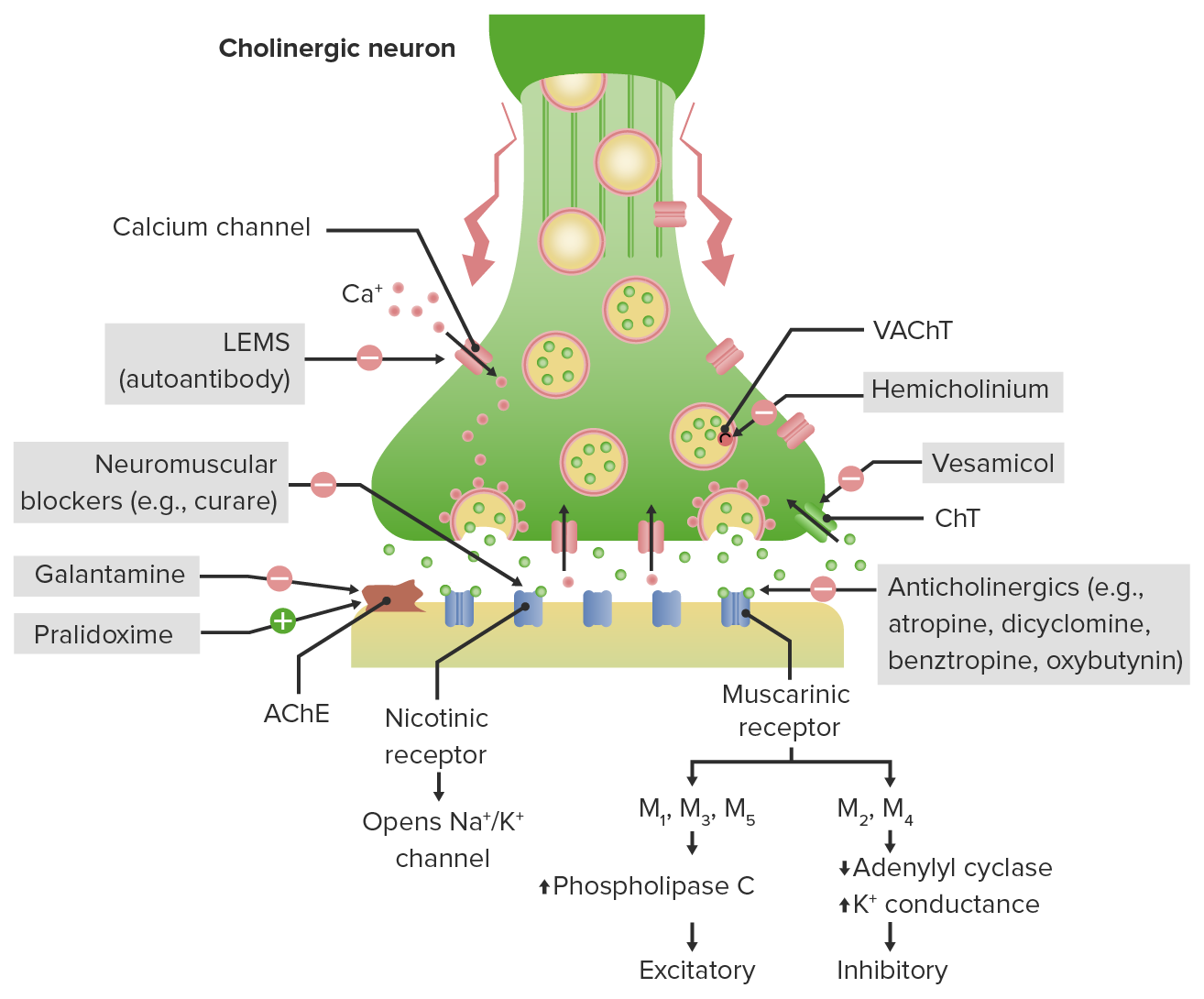
Cholinergic terminal neurotransmission and mechanism of action of anticholinergics:
Acetylcholine (ACh) is terminated in the synaptic cleft by AChE (acetylcholinesterase).
Anticholinergic drugs antagonize different muscarinic receptors throughout the body depending on their selectivity.
LEMS (Lambert-Eaton myasthenic syndrome) produces antibodies that block presynaptic calcium channels → decreased ACh release and muscle weakness.
Neuromuscular blockers antagonize nicotinic receptors at neuromuscular junctions.
Galantamine: AChE inhibitor used in Alzheimer dementia; inhibits the breakdown of ACh for a net increase in ACh in the synaptic cleft
Pralidoxime (2-PAM): AChE regenerator, allows ↑ breakdown of ACh → net decrease in ACh in the synaptic cleft.
CHT: choline transporter
VAChT: vesicular acetylcholine transporter
The pharmacokinetic properties of anticholinergic drugs are diverse, as they are available in many forms for different medical uses, such as IV atropine for severe bradycardia Bradycardia Bradyarrhythmia is a rhythm in which the heart rate is less than 60/min. Bradyarrhythmia can be physiologic, without symptoms or hemodynamic change. Pathologic bradyarrhythmia results in reduced cardiac output and hemodynamic instability causing syncope, dizziness, or dyspnea. Bradyarrhythmias, oral tablets for GI and bladder Bladder A musculomembranous sac along the urinary tract. Urine flows from the kidneys into the bladder via the ureters, and is held there until urination. Pyelonephritis and Perinephric Abscess antispasmodic Antispasmodic Antispasmodics are a group of medications used to reduce excessive GI smooth muscle contractility and spasm. These medications may be helpful in those with abdominal pain due to conditions such as irritable bowel syndrome, although their efficacy is controversial. Antispasmodics use, inhaled forms of LAMAs for bronchodilation, and eye drops for cycloplegia.
| Drug | Maximum mydriasis Mydriasis Dilation of pupils to greater than 6 mm combined with failure of the pupils to constrict when stimulated with light. This condition may occur due to injury of the pupillary fibers in the oculomotor nerve, in acute angle-closure glaucoma, and in adie syndrome. Glaucoma | Duration of cycloplegia |
|---|---|---|
| Atropine | 30–40 minutes | 7–12 days |
| Cyclopentolate ophthalmic | 30 minutes | 24 hours |
| Homatropine | 40–60 minutes | 24 hours |
| Tropicamide | 30 minutes | 6 hours |
| Drug | Half-life Half-Life The time it takes for a substance (drug, radioactive nuclide, or other) to lose half of its pharmacologic, physiologic, or radiologic activity. Pharmacokinetics and Pharmacodynamics | Lipophilicity (CNS penetration Penetration X-rays) | Metabolism |
|---|---|---|---|
| Darifenacin (Enablex) | 13–19 hours | Moderate |
|
| Fesoterodine (Toviaz) | 7 hours | Low |
|
| Oxybutynin (Ditropan, Ditropan XL, Oxytrol patch Patch Nonpalpable lesion > 1 cm in diameter Generalized and Localized Rashes, Gelnique topical gel) |
|
Lipophilic |
|
| Solifenacin (Vesicare) | 45–68 hours | Lipophilic | Hepatic CYP3A4 CYP3A4 Class 3 Antiarrhythmic Drugs (Potassium Channel Blockers) |
| Tolterodine (Detrol IR, LA, and ER forms) |
|
Low |
|
| Trospium (Sanctura, also XR form) | 20–35 hours | None | Hepatic |
Anticholinergic drugs are used in many forms. They are used via eye drops in ophthalmology as mydriatics/cycloplegics, included in inhalers as bronchodilators Bronchodilators Asthma Drugs and to dry up secretions as LAMAs for individuals with COPD COPD Chronic obstructive pulmonary disease (COPD) is a lung disease characterized by progressive, largely irreversible airflow obstruction. The condition usually presents in middle-aged or elderly persons with a history of cigarette smoking. Signs and symptoms include prolonged expiration, wheezing, diminished breath sounds, progressive dyspnea, and chronic cough. Chronic Obstructive Pulmonary Disease (COPD), and used in oral pill form as antispasmodics. Atropine is also used IV for severe bradycardia Bradycardia Bradyarrhythmia is a rhythm in which the heart rate is less than 60/min. Bradyarrhythmia can be physiologic, without symptoms or hemodynamic change. Pathologic bradyarrhythmia results in reduced cardiac output and hemodynamic instability causing syncope, dizziness, or dyspnea. Bradyarrhythmias and is part of the ACLS protocol.
Adverse effects are due to effects on the CNS and also to reduced activity of the parasympathetic nervous system Nervous system The nervous system is a small and complex system that consists of an intricate network of neural cells (or neurons) and even more glial cells (for support and insulation). It is divided according to its anatomical components as well as its functional characteristics. The brain and spinal cord are referred to as the central nervous system, and the branches of nerves from these structures are referred to as the peripheral nervous system. Nervous System: Anatomy, Structure, and Classification. Anticholinergics must be used with caution or avoided in the elderly owing to the risks of side effects.
| Adverse effects | Warnings/ contraindications Contraindications A condition or factor associated with a recipient that makes the use of a drug, procedure, or physical agent improper or inadvisable. Contraindications may be absolute (life threatening) or relative (higher risk of complications in which benefits may outweigh risks). Noninvasive Ventilation | Drug–drug interactions | |
|---|---|---|---|
| Atropine |
|
Caution with:
|
Avoid with:
|
| GI antispasmodics: Dicyclomine and Hyoscyamine | Antimuscarinic effects similar to atropine | Warnings similar to atropine | Similar to atropine drug interactions |
Urinary antispasmodics:
|
|
|
|
A cholinergic crisis Cholinergic Crisis Myasthenia Gravis may occur with an overdose or with using multiple drugs that have cholinergic effects.
| Group | Drug | Mechanism of action | Indications |
|---|---|---|---|
| Antimuscarinic agents | Atropine |
|
|
| Glycopyrrolate | Antagonizes ACh ACh A neurotransmitter found at neuromuscular junctions, autonomic ganglia, parasympathetic effector junctions, a subset of sympathetic effector junctions, and at many sites in the central nervous system. Receptors and Neurotransmitters of the CNS receptors Receptors Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors (anticholinergic) |
|
|
| Antiparkinson drugs Antiparkinson drugs Medications for the management of Parkinson’s disease improve the symptoms of tremor, rigidity, and postural instability by increasing dopamine levels in the brain. While levodopa is the drug of choice in individuals of any age with moderate or severe symptoms, other agents can be used as monotherapy for milder symptoms or in conjunction with levodopa–carbidopa for symptom control. Parkinson’s Disease Drugs |
|
Antagonize muscarinic receptors Muscarinic Receptors Asthma Drugs in the striatum Striatum Striped gray matter and white matter consisting of the neostriatum and paleostriatum (globus pallidus). It is located in front of and lateral to the thalamus in each cerebral hemisphere. The gray substance is made up of the caudate nucleus and the lentiform nucleus (the latter consisting of the globus pallidus and putamen). The white matter is the internal capsule. Basal Ganglia: Anatomy |
|
| Antiemetics Antiemetics Antiemetics are medications used to treat and/or prevent nausea and vomiting. These drugs act on different target receptors. The main classes include benzodiazepines, corticosteroids, atypical antipsychotics, cannabinoids, and antagonists of the following receptors: serotonin, dopamine, and muscarinic and neurokinin receptors. Antiemetics | Scopolamine Scopolamine An alkaloid from solanaceae, especially datura and scopolia. Scopolamine and its quaternary derivatives act as antimuscarinics like atropine, but may have more central nervous system effects. Its many uses include an anesthetic premedication, the treatment of urinary incontinence and motion sickness, an antispasmodic, and a mydriatic and cycloplegic. Antiemetics ( patch Patch Nonpalpable lesion > 1 cm in diameter Generalized and Localized Rashes) | Nonselective muscarinic receptor Receptor Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors antagonism |
|
| GI antispasmodics | Dicyclomine | Antagonism of muscarinic receptors Muscarinic Receptors Asthma Drugs → relaxation of GI smooth muscle and ↓ GI motility GI Motility The primary functions of the GI tract are digestion and absorption, which require coordinated contractions of the smooth muscles present in the GI tract. Peristaltic waves, segmentation contractions, and the migrating motor complex are all important contraction patterns that help to mix contents, get them in contact with the intestinal walls, and propel material down the tract at appropriate times and in appropriate amounts. Gastrointestinal Motility | IBS IBS Irritable bowel syndrome (IBS) is a functional bowel disease characterized by chronic abdominal pain and altered bowel habits without an identifiable organic cause. The etiology and pathophysiology of this disease are not well understood, and there are many factors that may contribute. Irritable Bowel Syndrome |
| Hyoscyamine | Nonselective muscarinic
receptor
Receptor
Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.
Receptors antagonist—blocks
ACh
ACh
A neurotransmitter found at neuromuscular junctions, autonomic ganglia, parasympathetic effector junctions, a subset of sympathetic effector junctions, and at many sites in the central nervous system.
Receptors and Neurotransmitters of the CNS at postganglionic muscarinic
acetylcholine
Acetylcholine
A neurotransmitter found at neuromuscular junctions, autonomic ganglia, parasympathetic effector junctions, a subset of sympathetic effector junctions, and at many sites in the central nervous system.
Receptors and Neurotransmitters of the CNS
receptors
Receptors
Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.
Receptors (mAChRs) at:
|
|
|
| Urinary antispasmodics |
|
|
OAB |
| Cycloplegics/mydriatics (ophthalmic solutions) |
|
|
|
| Bronchodilators Bronchodilators Asthma Drugs (oral inhalation) |
|
|
COPD COPD Chronic obstructive pulmonary disease (COPD) is a lung disease characterized by progressive, largely irreversible airflow obstruction. The condition usually presents in middle-aged or elderly persons with a history of cigarette smoking. Signs and symptoms include prolonged expiration, wheezing, diminished breath sounds, progressive dyspnea, and chronic cough. Chronic Obstructive Pulmonary Disease (COPD) |
| AChE regenerator | Pralidoxime Pralidoxime Various salts of a quaternary ammonium oxime that reconstitute inactivated acetylcholinesterase, especially at the neuromuscular junction, and may cause neuromuscular blockade. They are used as antidotes to organophosphorus poisoning as chlorides, iodides, methanesulfonates (mesylates), or other salts. General Principles of Toxidromes |
|
Antidote Antidote An antidote is a substance that counteracts poisoning or toxicity. Substances that can cause poisoning include heavy metals (from occupation, treatments, or diet), alcohols, environmental toxins, and medications. Antidotes of Common Poisonings for nerve agent or organophosphate insecticide poisoning Insecticide Poisoning Insecticides are chemical substances used to kill or control insects, to improve crop yields, and to prevent diseases. Human exposures to insecticides can be by direct contact, inhalation, or ingestion. Important insecticides that can affect humans include organochlorines (dichlorodiphenyltrichloroethane (DDT)), organophosphates (malathion and parathion), and carbamates (carbaryl, propoxur, aldicarb, and methomyl). Insecticide Poisoning |