Hereditary spherocytosis ( HS HS Hypertrophic scars and keloids are raised, red, and rigid (3 rs) scars that develop during cutaneous wound healing and are characterized by a local abnormal proliferation of fibroblasts with over-production of collagen. Over-expression of growth factors and decreased production of molecules that promote matrix breakdown appear to be involved in the etiology. Hypertrophic and Keloid Scars) is the most common type of hereditary hemolytic anemia Hemolytic Anemia Hemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia. The condition is caused by a cytoskeletal protein deficiency in the RBC membrane. This results in loss of membrane stability and deformability of the RBC, giving the cell its spherical shape (spherocyte). These cells are predisposed to splenic degradation, leading to hemolysis. Examination may show jaundice Jaundice Jaundice is the abnormal yellowing of the skin and/or sclera caused by the accumulation of bilirubin. Hyperbilirubinemia is caused by either an increase in bilirubin production or a decrease in the hepatic uptake, conjugation, or excretion of bilirubin. Jaundice and splenomegaly Splenomegaly Splenomegaly is pathologic enlargement of the spleen that is attributable to numerous causes, including infections, hemoglobinopathies, infiltrative processes, and outflow obstruction of the portal vein. Splenomegaly, while laboratory tests are consistent with hemolytic anemia Hemolytic Anemia Hemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia and increased hemoglobin concentration. Among multiple confirmatory tests for HS HS Hypertrophic scars and keloids are raised, red, and rigid (3 rs) scars that develop during cutaneous wound healing and are characterized by a local abnormal proliferation of fibroblasts with over-production of collagen. Over-expression of growth factors and decreased production of molecules that promote matrix breakdown appear to be involved in the etiology. Hypertrophic and Keloid Scars, the eosin-5’-maleimide (EMA) binding test is preferred. The only definitive treatment for HS HS Hypertrophic scars and keloids are raised, red, and rigid (3 rs) scars that develop during cutaneous wound healing and are characterized by a local abnormal proliferation of fibroblasts with over-production of collagen. Over-expression of growth factors and decreased production of molecules that promote matrix breakdown appear to be involved in the etiology. Hypertrophic and Keloid Scars is splenectomy Splenectomy Surgical procedure involving either partial or entire removal of the spleen. Rupture of the Spleen.
Last updated: Dec 15, 2025
In HS HS Hypertrophic scars and keloids are raised, red, and rigid (3 rs) scars that develop during cutaneous wound healing and are characterized by a local abnormal proliferation of fibroblasts with over-production of collagen. Over-expression of growth factors and decreased production of molecules that promote matrix breakdown appear to be involved in the etiology. Hypertrophic and Keloid Scars, gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics mutations lead to cytoskeletal protein deficiency:
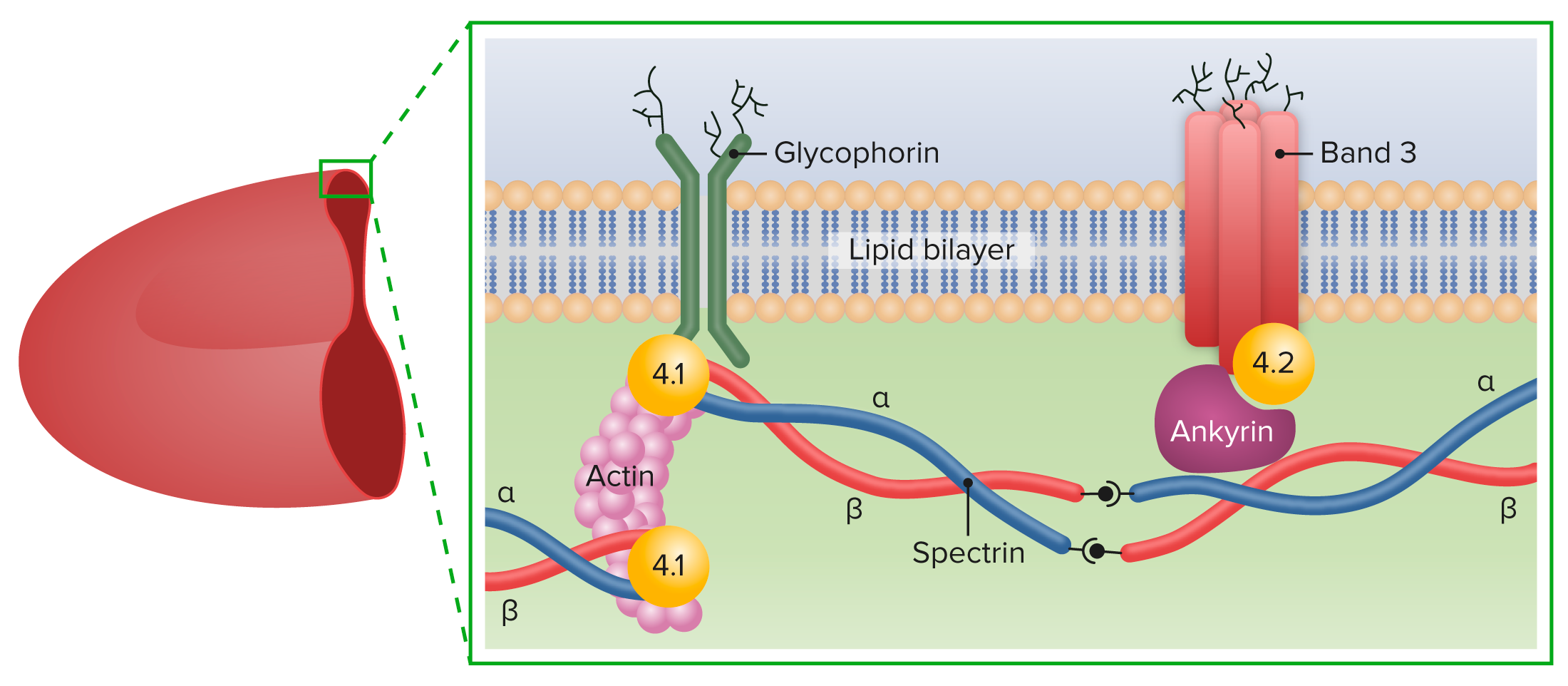
Major proteins of the RBC membrane
Image by Lecturio.Four clinical categories have been established based on hemoglobin ( Hb Hb The oxygen-carrying proteins of erythrocytes. They are found in all vertebrates and some invertebrates. The number of globin subunits in the hemoglobin quaternary structure differs between species. Structures range from monomeric to a variety of multimeric arrangements. Gas Exchange), reticulocyte Reticulocyte Immature erythrocytes. In humans, these are erythroid cells that have just undergone extrusion of their cell nucleus. They still contain some organelles that gradually decrease in number as the cells mature. Ribosomes are last to disappear. Certain staining techniques cause components of the ribosomes to precipitate into characteristic ‘reticulum’ (not the same as the endoplasmic reticulum), hence the name reticulocytes. Erythrocytes: Histology (retic) count, and bilirubin Bilirubin A bile pigment that is a degradation product of heme. Heme Metabolism (bili) level.
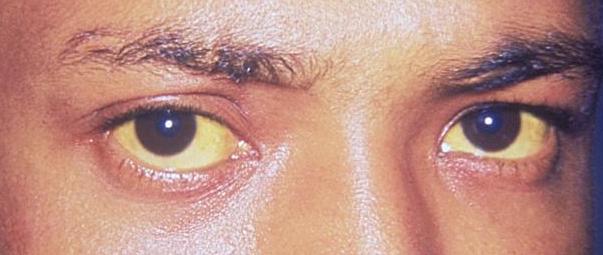
Scleral icterus: The 1st clinical sign of bilirubin deposition in the body
Image: “Jaundice eye new” by CDC/Dr. Thomas F. Sellers/Emory University. License: Public Domain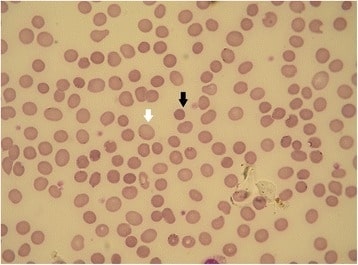
Hereditary spherocytosis (HS): peripheral blood smear
The black arrow shows a spherocyte. The white arrow shows a normal RBC (the lack of central pallor is an artifact). Hereditary spherocytosis forms having different membrane defects can show different red cell morphologies.
The differential diagnosis includes other causes of hemolysis, most of which will show at least some spherocytes in the peripheral blood.