


A esferocitose hereditária (EH) é o tipo mais MAIS Androgen Insensitivity Syndrome comum de anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica hereditária. Esta condição é causada por uma deficiência de uma proteína do citoesqueleto na membrana dos glóbulos vermelhos (GV). Isso resulta na perda de estabilidade e deformabilidade da membrana dos GV, dando à célula uma forma esférica (esferócito). Essas células estão predispostas à degradação esplênica, levando à hemólise. O exame físico pode mostrar icterícia e esplenomegalia, enquanto os exames laboratoriais são consistentes com anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica e aumento da concentração de hemoglobina. Entre os múltiplos testes Testes Gonadal Hormones confirmatórios para EH, o teste de ligação da eosina-5'-maleimida (EMA) é o preferido. O único tratamento definitivo para EH é a esplenectomia.
Last updated: Dec 15, 2025
Na EH, as mutações genéticas levam à deficiência da proteína do citoesqueleto :
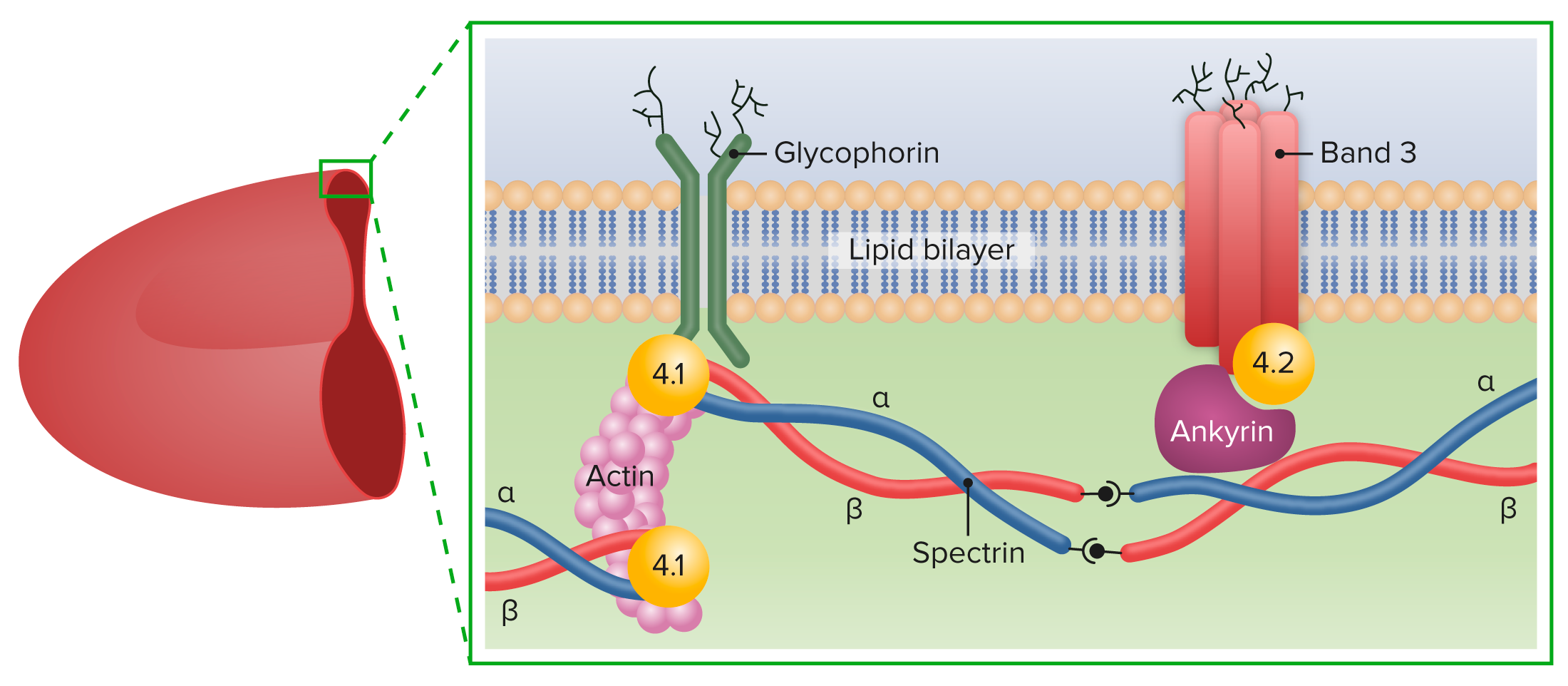
Principais proteínas da membrana dos GV
Imagem de Lecturio.Quatro categorias de apresentações clínicas foram estabelecidas com base na hemoglobina ( Hb Hb The oxygen-carrying proteins of erythrocytes. They are found in all vertebrates and some invertebrates. The number of globin subunits in the hemoglobin quaternary structure differs between species. Structures range from monomeric to a variety of multimeric arrangements. Gas Exchange), contagem de reticulócitos (retic) e nível de bilirrubina (bili).
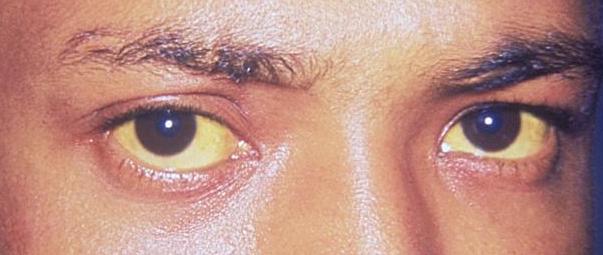
Icterícia das escleróticas: o primeiro sinal clínico de deposição de bilirrubina no organismo
Imagem: “Jaundice eye new” de CDC / Dr. Thomas F. Sellers / Emory University. Licença: Domínio Público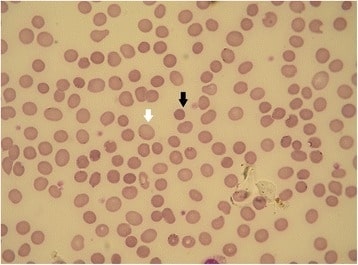
Esferocitose hereditária (EH): esfregaço de sangue periférico
A seta preta mostra um esferócito. A seta branca mostra um GV normal (a falta de palidez central é um artefato). As formas de esferocitose hereditária com diferentes defeitos de membrana podem apresentar diferentes morfologias de GV.
O diagnóstico diferencial inclui outras causas de hemólise, a maioria das quais mostrará pelo menos alguns esferócitos no sangue periférico.
