Hypoplastic left heart syndrome (HLHS) is a congenital heart defect that consists of the underdevelopment, or hypoplasia, of the left side of the heart in various degrees. The most notable feature of HLHS is the reduced size and functionality of the left ventricle (LV). Also, HLHS is associated with stenosis, hypoplasia, or atresia of the vessels or atrioventricular valves on the left side of the heart. A mixture of genetic factors and altered fetal blood flow Blood flow Blood flow refers to the movement of a certain volume of blood through the vasculature over a given unit of time (e.g., mL per minute). Vascular Resistance, Flow, and Mean Arterial Pressure causes HLHS. Hypoplastic left heart syndrome presents once the ductus arteriosus Ductus arteriosus A fetal blood vessel connecting the pulmonary artery with the descending aorta. Patent Ductus Arteriosus (PDA) closes physiologically as tachypnea Tachypnea Increased respiratory rate. Pulmonary Examination, cyanosis Cyanosis A bluish or purplish discoloration of the skin and mucous membranes due to an increase in the amount of deoxygenated hemoglobin in the blood or a structural defect in the hemoglobin molecule. Pulmonary Examination, heart failure Heart Failure A heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction. Total Anomalous Pulmonary Venous Return (TAPVR), and cardiogenic shock Cardiogenic shock Shock resulting from diminution of cardiac output in heart disease. Types of Shock. Diagnosis can be made pre- or postnatally via echocardiogram Echocardiogram Transposition of the Great Arteries. Once detected, surgical treatment is the 1st-line therapy, done in 3 stages.
Last updated: Feb 16, 2026
Hypoplastic left heart syndrome (HLHS) is characterized by the underdevelopment of the left side of the heart. The components of HLHS include:
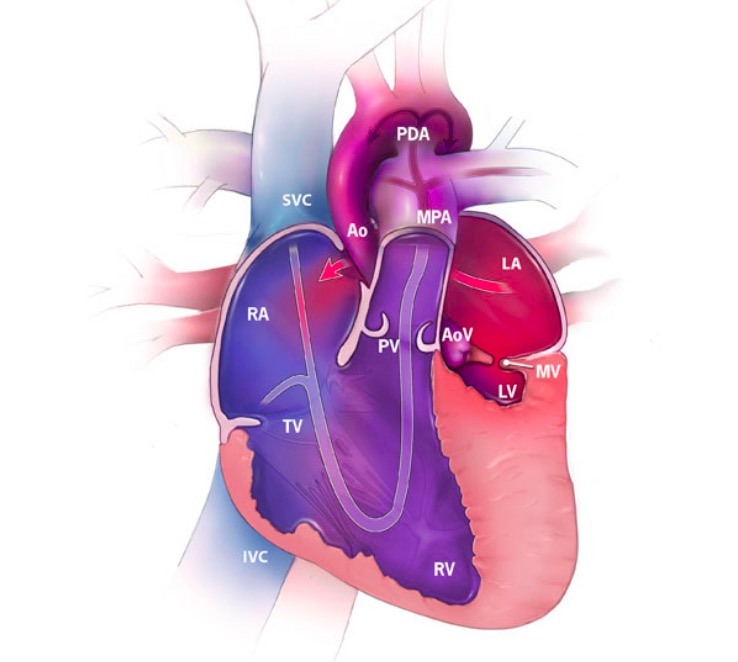
Hypoplastic left heart syndrome (HLHS) featuring the direction of blood flow (arrows) and the oxygenation of blood:
Red represents oxygenated blood, blue shows deoxygenated blood, and purple shows mixed blood.
RA: right atrium
RV: right ventricle
LA: left atrium
LV: left ventricle
SVC: superior vena cava
IVC: inferior vena cava
MPA: main pulmonary artery
Ao: aorta
PDA: patent ductus arteriosus
TV: tricuspid valve
MV: mitral valve
PV: pulmonary valve
AoV: aortic valve
There are 3 types of HLHS based on the morphology of the cardiac valves:
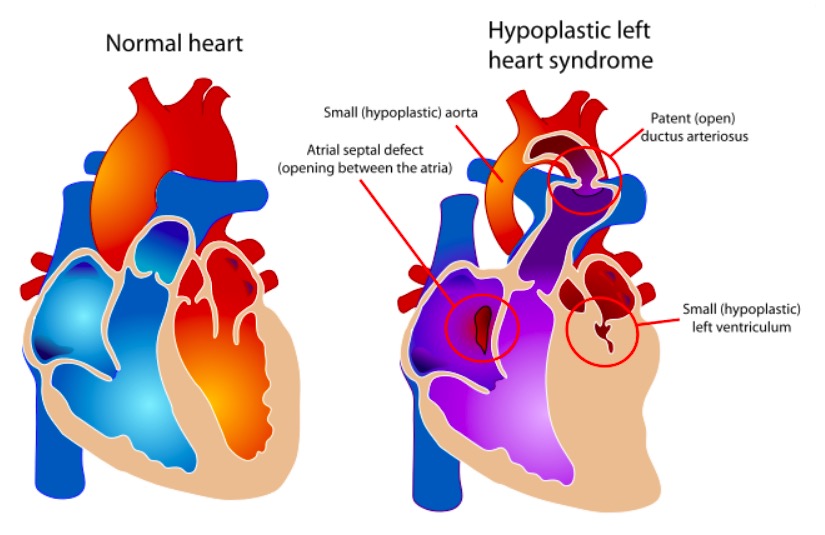
The components of HLHS vs. a normal heart
Image: “Hypoplastic left heart syndrome” by Mariana Ruiz. License: Public Domain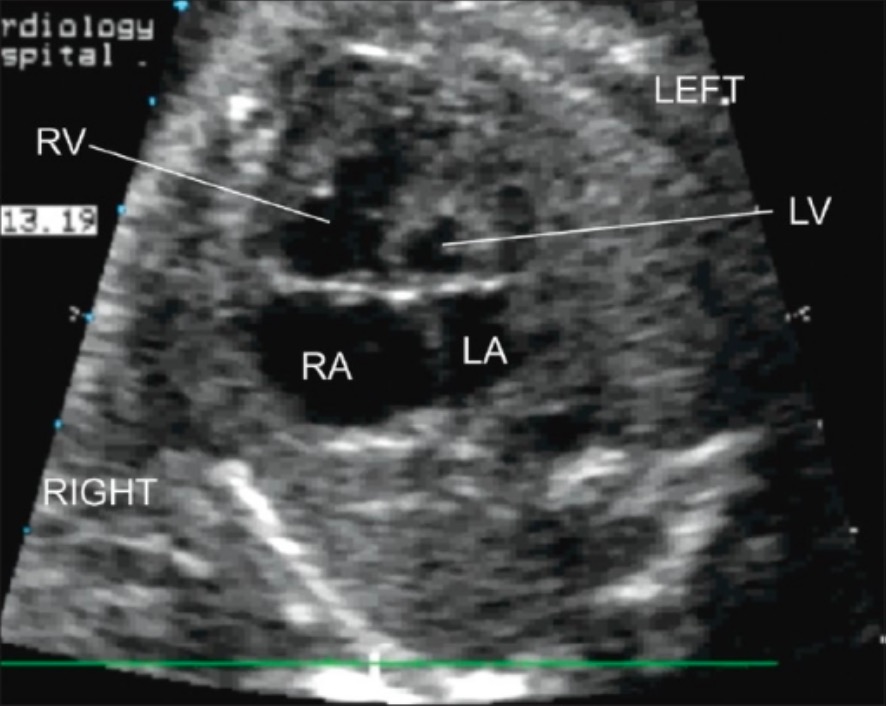
Postnatal transthoracic echocardiography (4-chamber view):
Severe hypoplasia of the LV with hypoplasia of the LA are seen.
LA: left atrium
LV: left ventricle
RA: right atrium
RV: right ventricle
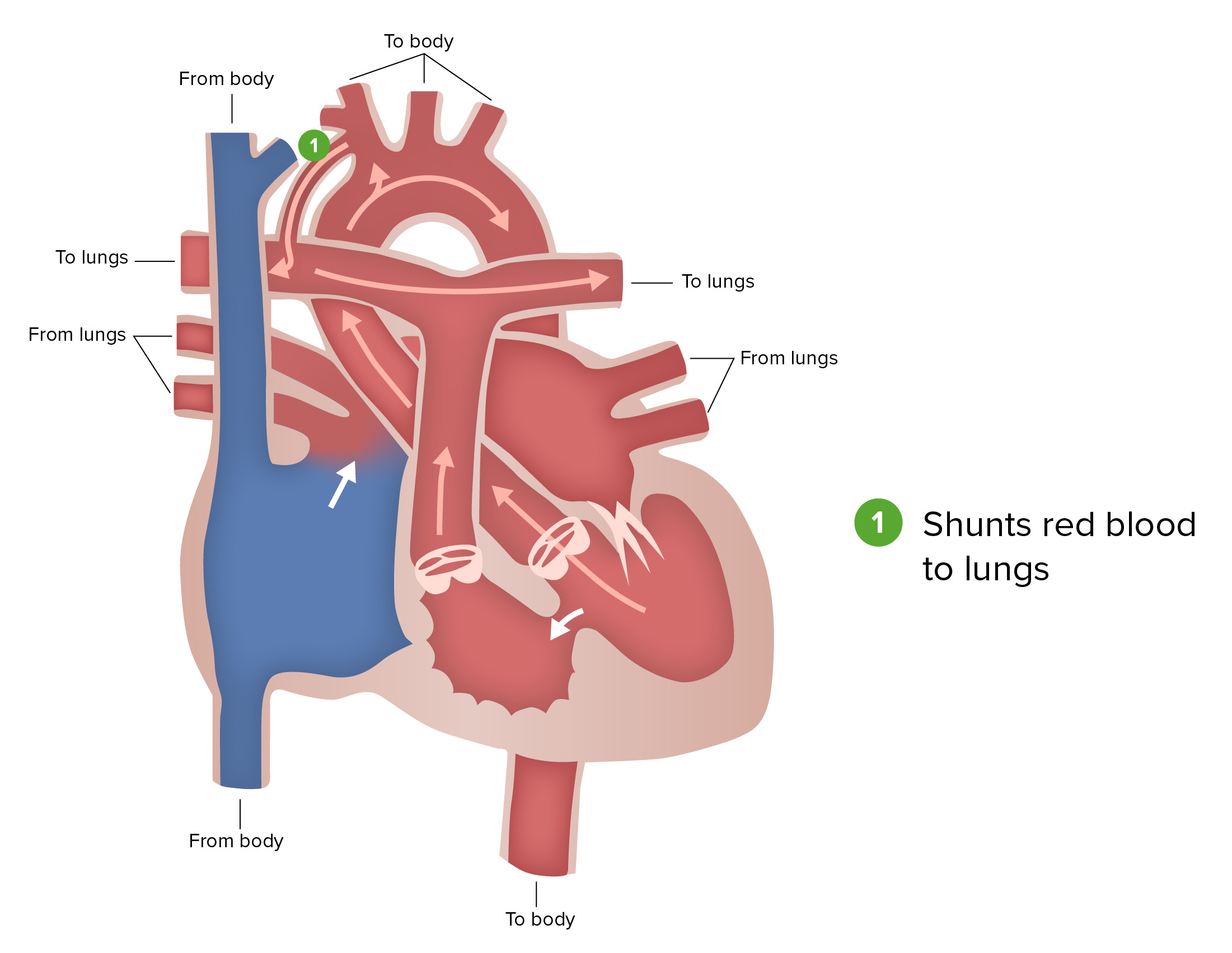
Blalock-Taussig shunt:
The Blalock-Taussig shunt, a part of the first stage of surgical repair performed for patients with hypoplastic left heart syndrome, involves connecting a branch of the subclavian or carotid artery to the pulmonary artery.
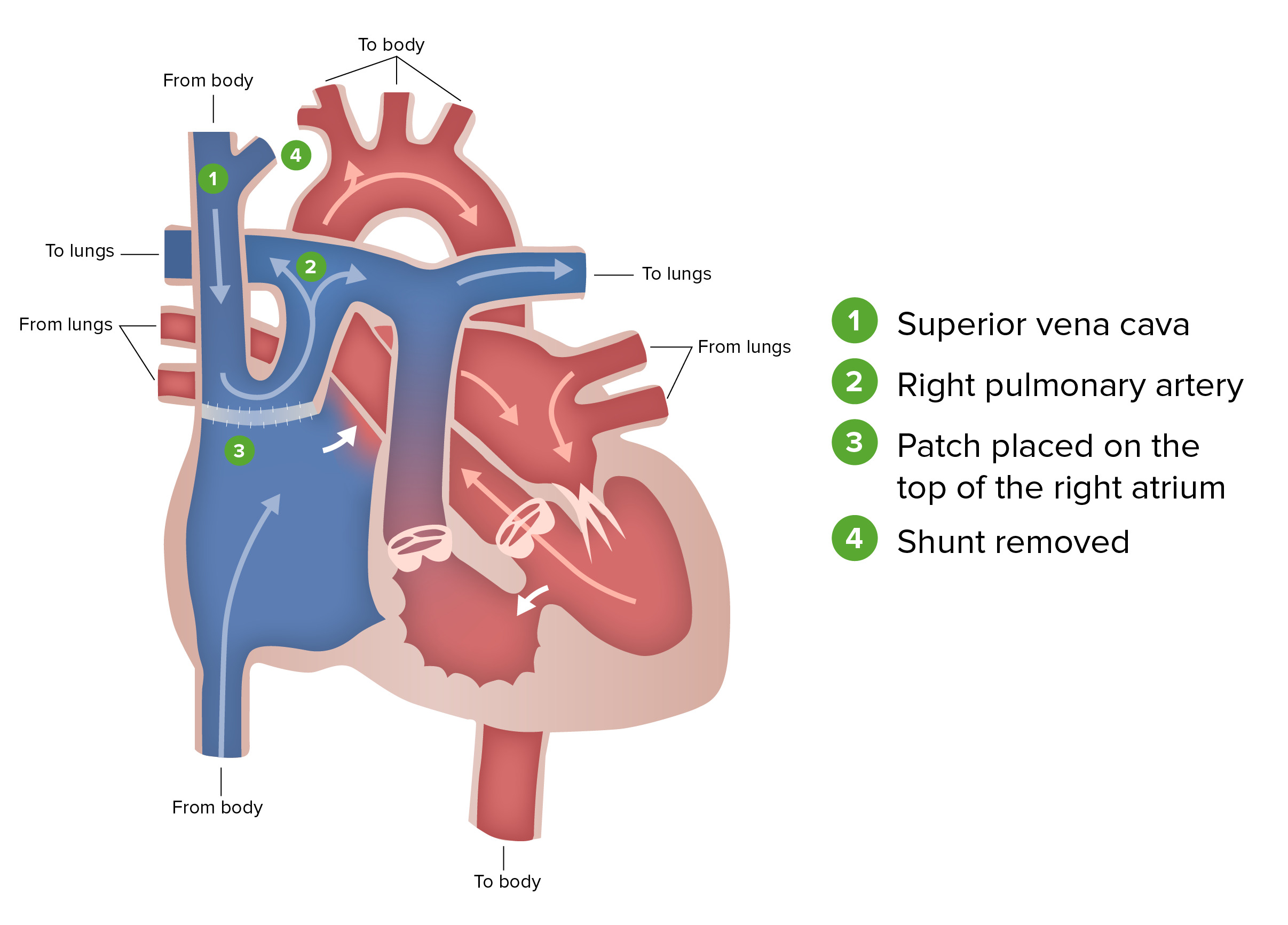
Glenn procedure:
The Glenn procedure, the second stage of surgical repair performed for patients with hypoplastic left heart syndrome, involves ligating the superior vena cava from the heart and connecting it to the pulmonary circulation. During this stage, the Blalock-Taussig shunt is removed.

Fontan procedure:
The Fontan procedure, the final stage of surgical repair performed for patients with hypoplastic left heart syndrome, involves the redirection of venous blood from the lower body, through the inferior vena cava and into the pulmonary artery.
The following genetic syndromes are associated with the development of HLHS:
The following conditions are differential diagnoses of HLHS: