
Start your online cardiology class
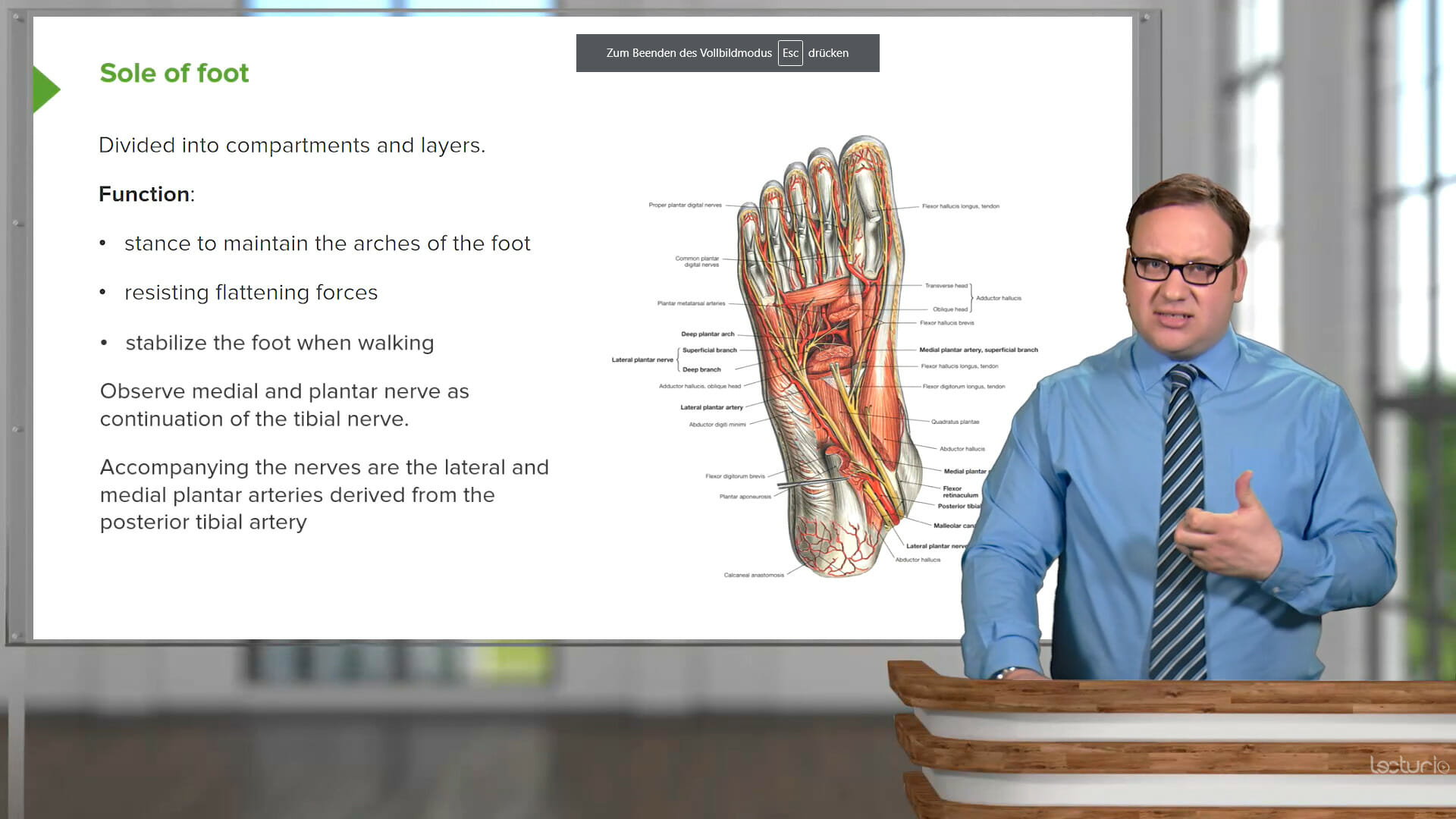
with by Dr. Joseph Alpert, MD, from Tucson University, Arizona
Cardiovascular disease is one of the major public health concerns in the western world. Its associated morbidity can severely impair patients’ function and quality of life. The intricacies of diagnosing and treating conditions like heart failure, valvular diseases, and coronary artery disease are challenging.
This course provides an in-depth look into the causes, signs, diagnosis, and treatment of major cardiac diseases, including heart failure, valvular diseases, conduction abnormalities (bradyarrhythmias and tachyarrhythmias), congenital abnormalities, and particularly coronary artery disease. You will learn how to recognize common signs and symptoms of cardiac anomalies and become proficient in analyzing ECG tracings. Your educator, Dr. Joseph Alpert, is an experienced cardiologist who shares his extensive expertise to help you gain practical, real-world knowledge that extends beyond textbook theories.
The combination of Video Lessons with interactive quiz questions, downloadable study materials, and a USMLE-style Qbank makes it easy to understand and retain the topics. By the end of this course, you will have a thorough understanding of cardiology, ready to diagnose and treat various cardiac conditions effectively, and prepared to excel in your medical exams and clinical practice.