Los LOS Neisseria tumores neuroendocrinos pancreáticos (PanNET, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) surgen del páncreas endocrino (células de los LOS Neisseria islotes) y representan entre el 1% y el 2% de las neoplasias pancreáticas primarias; el >90% restante proviene del páncreas exocrino. La mayoría de los LOS Neisseria PanNET son no funcionales (50%-75%), mientras que la minoría funcional puede ser benigna o maligna. Los LOS Neisseria PanNET benignos o malignos incluyen insulinomas Insulinomas An insulinoma is a type of functional neuroendocrine tumor that manifests with hypoglycemia due to autologous secretion of insulin. It more commonly presents as a solitary benign tumor but can sometimes be associated with MEN type 1 (MEN1). Patients present with fasting hypoglycemia, which may manifest as episodes of diaphoresis, palpitations, tremor, and confusion. Insulinomas, gastrinomas, carcinomas neuroendocrinos pancreáticos poco diferenciados y tumores muy raros llamados glucagonomas, somatostatinomas y vipomas, que reciben su nombre de las hormonas que secretan. El diagnóstico se realiza clínicamente, con pruebas de laboratorio para la secreción hormonal, así como imágenes o endoscopia digestiva alta. El tratamiento es quirúrgico y puede incluir análogos de la somatostatina, fármacos dirigidos como everolimus Everolimus A derivative of sirolimus and an inhibitor of tor serine-threonine kinases. It is used to prevent graft rejection in heart and kidney transplant patients by blocking cell proliferation signals. It is also an antineoplastic agent. Immunosuppressants o sunitinib, y terapia con radionúclidos de receptores peptídicos (PRRT).
Last updated: Dec 15, 2025
3 de las mutaciones somáticas más comunes:
Los LOS Neisseria pacientes pueden presentar una enfermedad metastásica hepática, que se confirma con una biopsia. El enfoque de diagnóstico se centra en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la identificación tanto de la extensión de la enfermedad como del sitio primario probable de la misma.
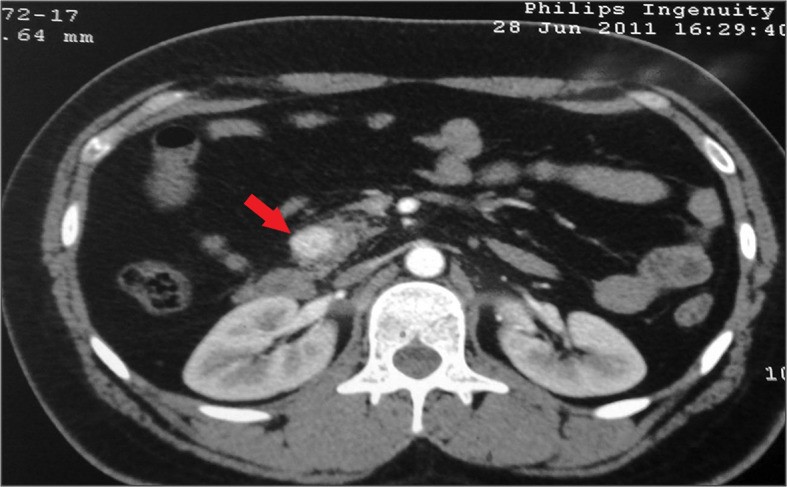
TC con contraste (sección transversal) que muestra un insulinoma solitario en la cabeza del páncreas (flecha)
Imagen: “Insulinoma presenting as refractory seizure disorder” por Correia P, Panchani R, Ranjan R, Agrawal C. Licencia: CC BY 3.0, editado por Lecturio.En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum general, el tratamiento es quirúrgico si la neoplasia es resecable.