A acidose tubular renal (ATR) é um desequilíbrio no pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance fisiológico, causado pela incapacidade do rim em acidificar a urina e manter o pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance do sangue em níveis fisiológicos. A acidose tubular renal existe em vários tipos, incluindo ATR distal (tipo 1), ATR proximal (tipo 2), ATR mista (tipo 3) e ATR hipercalémica (tipo 4). Dependendo do tipo de ATR, existem vários mecanismos a causar disfunção da regulação ácido-base renal, resultando em acidose metabólica sem gap aniónico. Todas as ATRs apresentam, clinicamente, algum grau de acidose metabólica; no entanto, a ATR distal e a ATR proximal também apresentam hipocalémia, enquanto que a ATR hipercalémica não. O diagnóstico é estabelecido, sobretudo, com base na história e nas análises laboratoriais, incluindo a medição dos gaps aniónicos sérico e urinário. O tratamento envolve a correção da acidose metabólica crónica com agentes alcalinizantes, para prevenir os seus efeitos catabólicos a longo prazo nos ossos e músculos, bem como a abordagem das causas subjacentes que levam a esta condição.
A acidose tubular renal (ATR) é um desequilíbrio no pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance fisiológico, causado pela incapacidade do rim em acidificar a urina e manter o pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance do sangue em níveis fisiológicos.
Classificação
A ATR pode ser classificada com base nas características clínicas e no defeito fisiológico:
Tipo 1: ATR distal
Tipo 2: ATR proximal
Tipo 3: ATR mista (muito rara)
Tipo 4: ATR hipercalémica
Comparação dos tipos de ATR, incluindo as características clínicas, defeitos fisiológicos e potenciais etiologias:
Tabela: ATR tipo 1 (distal) – secreção ácida prejudicada
Características
Secreção de H+ prejudicada nos segmentos distais
pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance da urina > 5,3
PlasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products HCO3– variável
Defeito renal
↑ H+–K+–ATPase
↑ Permeabilidade tubular, permitindo refluxo de H+
↓ Reabsorção Na+
Etiologia
Doenças autoimunes familiares (síndrome de Sjögren, artrite reumatoide)
Fármacos, toxinas
Tabela: ATR tipo 2 (proximal) – secreção de bicarbonato prejudicada
Características
Reabsorção proximal de HCO3 prejudicada
pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance da urina variável
PlasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products HCO3– 12–20 mMMMMultiple myeloma (MM) is a malignant condition of plasma cells (activated B lymphocytes) primarily seen in the elderly. Monoclonal proliferation of plasma cells results in cytokine-driven osteoclastic activity and excessive secretion of IgG antibodies.Multiple Myeloma/L
Defeito renal
Disfunção tubular inespecífica ou mutações em genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure envolvidos na reabsorção de HCO3–
Etiologia
Familiar
Síndrome de Fanconi
Fármacos, toxinas
Inibidores da anidrase carbónica
Tabela: ATR tipo 3 (mista)— combina o distal com o proximal
Características
Mutações hereditárias da anidrase carbónica tipo II
Muito rara
Apresenta-se na infância
Defeito renal
Mutações hereditárias da anidrase carbónica tipo II
Etiologia
Padrão hereditário autossómico recessivo
A maioria dos casos são reportados no Norte de África, em áreas com grande prevalência de consanguinidade
Tabela: ATR tipo 4 (hipoaldosteronismo) – secreção ácida prejudicada
Características
Alteração da libertação de aldosterona ou da sua resposta
pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance da urina < 5,3
PlasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products HCO3– > 17 mMMMMultiple myeloma (MM) is a malignant condition of plasma cells (activated B lymphocytes) primarily seen in the elderly. Monoclonal proliferation of plasma cells results in cytokine-driven osteoclastic activity and excessive secretion of IgG antibodies.Multiple Myeloma/L
Hipercalemia
Defeito renal
Reabsorção de Na+, via canal epitelial de Na+, prejudicada
Nefrocalcinose: Em casos de hipercalciúria são visíveis depósitos de cálcio renais. Esta condição pode surgir de múltiplas causas, incluindo hiperparatiroidismo, intoxicação por vitamina D e sarcoidose e pode resultar em acidose tubular renal tipo 1.
Causas autoimunes (especialmente síndrome de Sjögren)
Genética:
Autossómica dominante ou autossómica recessiva
Antiporte apical Na+/H+ das células tubulares proximais
Cotransportador basolateral de Na+/HCO3– de células tubulares proximais
Fármacos:
Metais pesados (chumbo, mercúrio)
Inibidores da anidrase carbónica (como a acetazolamida e o topiramato)
Aminoglicosídeos
Antirretrovirais (especificamente, tenofovirTenofovirAn adenine analog reverse transcriptase inhibitor with antiviral activity against HIV-1 and hepatitis b. It is used to treat HIV infections and chronic hepatitis b, in combination with other antiviral agents, due to the emergence of antiviral drug resistance when it is used alone.Anti-HIV Drugs)
Ifosfamida
Cisplatina, oxaliplatina
Ácido valpróico
Diversos:
Nefrite intersticial
Défice de vitamina D
Hiperparatiroidismo secundário
Transplante renal
Outras doenças associadas à síndrome de Fanconi:
Tirosinemia
Cistinose
GalactosemiaGalactosemiaGalactosemia is a disorder caused by defects in galactose metabolism. Galactosemia is an inherited, autosomal-recessive condition, which results in inadequate galactose processing and high blood levels of monosaccharide. The rare disorder often presents in infants with symptoms of lethargy, nausea, vomiting, diarrhea, and jaundice. Galactosemia
Intolerância hereditária à frutose
Doença de von Gierke (doença de armazenamento de glicogénio tipo I)
Inibidores da calcineurina (ciclosporina, tacrolimusTacrolimusA macrolide isolated from the culture broth of a strain of streptomyces tsukubaensis that has strong immunosuppressive activity in vivo and prevents the activation of T-lymphocytes in response to antigenic or mitogenic stimulation in vitro.Immunosuppressants)
Inibidores da enzima conversora de angiotensina (IECA)
Bloqueadores do recetor de angiotensina (ARA)
Inibidores da renina
Heparina
Trimetoprim
Pentamidina
Diversos:
Obstrução crónica do trato urinário
Insuficiência adrenal primária
Doença grave
Fisiopatologia
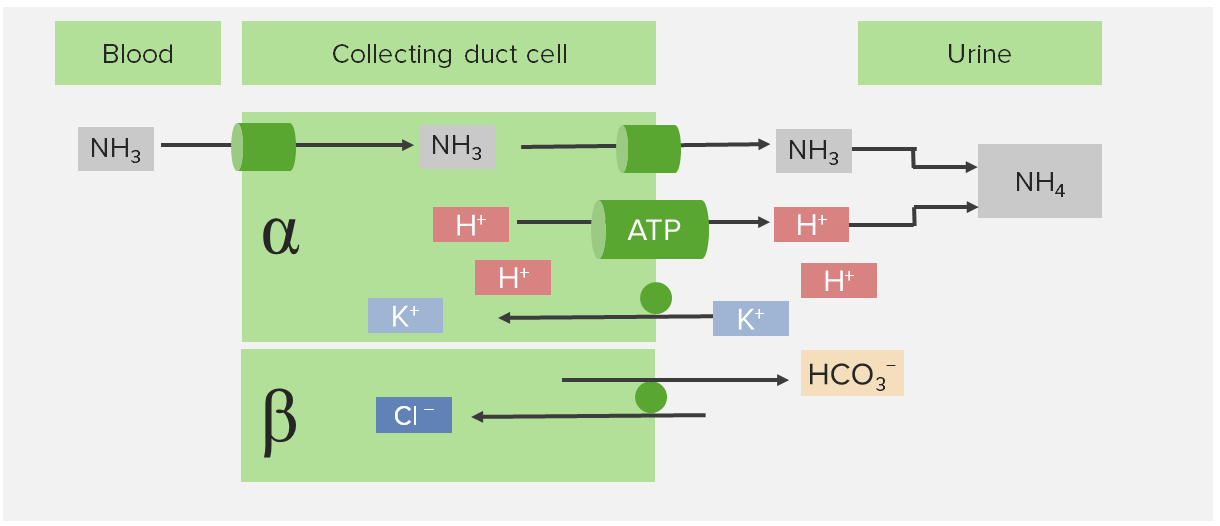
Acidose tubular renal distal (tipo 1)
A fisiopatologia da ATR distal (tipo 1) é a alteração da secreção ácida no ducto coletor do túbulo distal.
Processo normal de acidificação urinária:
As células intercaladas participam da excreção ácida:
Localizadas no ducto coletor do túbulo distal
Células α-intercaladas:
ATPase-H+/K+ apical (1 H+ para fora da célula, 1 K+ para dentro da célula)
ATPase-H+ apical (1 H+ para fora da célula)
ATPase-Na+/K+ basolateral
Trocador Cl–/HCO3– basolateral
Células β-intercaladas:
Trocador Cl – /HCO3– apical (1 HCO3– para fora da célula, 1 Cl– para dentro da célula)
ATPase-H+/K+ apical (1 H+ para fora da célula, 1 K+ para dentro da célula)
ATPase-Na+/K+ basolateral
ATPase-H+ basolateral
Efeito final
As células intercaladas secretam H+ no lúmen tubular
O H+ combina-se com NH3+ (amónia) e outros compostos (ácidos tituláveis)
São excretados NH4+ (amónio) e ácidos tituláveis.
A homeostase ácido-base é mantida
Processos anormais que levam à ATR distal:
↓ Atividade de ATPase-H+/K+ apical (↓ secreção H+ no lúmen tubular)
↑ secreção HCO3– → perda de HCO3– → acidose metabólica
Ocorre nas células β-intercaladas
↓ Atividade do trocador Cl–/HCO3– basolateral :
↓ secreção HCO3– → acumulação de HCO3– intracelular → ↑ pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance intracelular
Ocorre ↓ da secreção de H+ via ATPases apicais para corrigir o pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance intracelular.
Ocorre nas células α-intercaladas
Complicações:
Pode ocorrer acidose metabólica grave na ATR distal (tipo 1):
HCO3– sérico < 10 mEq/L se não tratada
Ocorre no nefrónio distal → não existem processos a jusante para compensar
Hipocalemia
Hipocitratúria (predispõe à nefrolitíase)
Células intercaladas na ATR distal (tipo I)
Imagem por Lecturio.
Acidose tubular renal proximal (tipo 2)
A fisiopatologia da ATR proximal (tipo II) baseia-se na alteração da reabsorção de bicarbonato no túbulo proximal.
Processo normal de reabsorção de HCO3–no túbulo proximal:
Em circunstâncias normais, 80% do HCO3– filtrado é reabsorvido no túbulo proximal.
Requer um mecanismo complexo porque o HCO3– não é livremente permeável (devido à carga):
O trocador 3 de iões sódio-hidrogénio (NHE3NHE3A sodium-hydrogen antiporter expressed primarily by epithelial cells in the kidneys, it localizes to the apical membrane of the proximal kidney tubule, where it functions in sodium and water reabsorption and possibly calcium homeostasis. It also is expressed in heart, brain, and lung tissues and is resistant to amiloride inhibition.Carbonic Anhydrase Inhibitors, pela sigla em inglês) absorve Na+ e secreta H+.
O H+ secretado combina-se com o HCO3– filtrado para formar H2CO3 no lúmen tubular.
O H2CO3 é convertido em H2O e CO2 pela anidrase carbónica apical IV.
O CO2 difunde-se livremente através da membrana apical, de volta para a célula.
A anidrase carbónica II intracelular converte CO2 + H2O novamente em H2CO3.
O H2CO3 pode, então, dissociar-se em H+ e HCO3–:
O H+ é reciclado em NHE3NHE3A sodium-hydrogen antiporter expressed primarily by epithelial cells in the kidneys, it localizes to the apical membrane of the proximal kidney tubule, where it functions in sodium and water reabsorption and possibly calcium homeostasis. It also is expressed in heart, brain, and lung tissues and is resistant to amiloride inhibition.Carbonic Anhydrase Inhibitors
O HCO3– é absorvido através da membrana basolateral via cotransportador Na+–HCO3– e trocador HCO3– –Cl–.
Resultados finais de todo o processo:
Excreção de H+
Absorção de HCO3–
Processos anormais que levam à acidose tubular renal proximal (tipo 2):
Mieloma múltiplo: as cadeias leves são diretamente tóxicas para as células do túbulo proximal
Fármacos que causam toxicidade às células do túbulo proximal através de múltiplos mecanismos
Mutações no:
NHE3NHE3A sodium-hydrogen antiporter expressed primarily by epithelial cells in the kidneys, it localizes to the apical membrane of the proximal kidney tubule, where it functions in sodium and water reabsorption and possibly calcium homeostasis. It also is expressed in heart, brain, and lung tissues and is resistant to amiloride inhibition.Carbonic Anhydrase Inhibitors apical
Cotransportador Na+/HCO3– basolateral
Complicações:
Acidose metabólica moderada
HCO3– sérico de 14–20 mEq/L, mesmo sem tratamento
A maioria da capacidade de reabsorção de HCO3– no túbulo proximal está mantida:
O limiar para a reabsorção de HCO3–, no túbulo proximal, está reduzido
HCO3– sérico ↓ até que atinja o novo limite (estado estacionário)
A acidificação distal está mantida → a carga ácida diária é controlada sem agravamento da acidose
Hipocalemia
Síndrome de Fanconi
Reabsorção de bicarbonato no túbulo proximal
CA-IV, pela sigla em inglês: anidrase carbónica IV CA-II, pela sigla em inglês: anidrase carbónica II
O defeito subjacente na ATR do tipo 3 consiste em anomalias tubulares renais proximais e distais, que resultam em acidémia sistémica grave.
Mutação ARARAortic regurgitation (AR) is a cardiac condition characterized by the backflow of blood from the aorta to the left ventricle during diastole. Aortic regurgitation is associated with an abnormal aortic valve and/or aortic root stemming from multiple causes, commonly rheumatic heart disease as well as congenital and degenerative valvular disorders. Aortic Regurgitation no geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of GeneticsCA2 no cromossoma 8q22:
Deficiência de anidrase carbónica II (CAII, pela sigla em inglês):
Defeito na reabsorção proximal de bicarbonato
Incapacidade de acidificar a urina, distalmente, no tubo coletor
Grave desperdício de bicarbonato:
Níveis de bicarbonato sérico perigosamente baixos
Desperdício renal concomitante de potássio
Hipocalemia:
Devido à falha da reabsorção de potássio, dependente da anidrase carbónica
Ocorre no nefrónio distal → nenhum processo a jusante para compensar
Acidose tubular renal hipercalémica (tipo 4)
O mecanismo clássico, na maioria dos indivíduos com ATR hipercalémica (ou seja, nefropatia diabética e DRC leve a moderada), éo hipoaldosteronismo hiporreninémico.
Ações normais da aldosterona:
O efeito final é:
Reabsorção de Na+
Secreção de K+
Secreção de H+
Estimula o canal epitelial de Na (ENaCENaCSodium channels found on salt-reabsorbing epithelial cells that line the distal nephron; the distal colon; salivary ducts; sweat glands; and the lung. They are amiloride-sensitive and play a critical role in the control of sodium balance, blood volume, and blood pressure.Liddle Syndrome, pela sigla em inglês) na célula principal
↑ Reabsorção de Na+ → ↑ gradiente elétrico para secreção de K+
A secreção de K+ ocorre através do canal de potássio medular externo (ROMK, pela sigla em inglês) nas células principais renais.
A hipercalemia contribui para a acidose metabólica:
O ↑ K+ inibe a amoniagénese no túbulo proximal
↓ do amónio urinário → ↓ excreção ácida
Todas as outras etiologias também envolvem algum distúrbio no SRAA → ↓ aldosterona, que pode ser absoluta ou relativa (ou seja, resistência à aldosterona).
As acidoses tubulares renais, muitas vezes, não têm uma apresentação clínica específica e só são consideradas quando a acidose metabólica é descoberta. Embora alguns indivíduos sejam assintomáticos, muitos apresentam sintomas significativos causados pela etiologia subjacente da ATR, em vez de sintomas da própria acidose.
Acidose tubular renal distal (tipo 1)
Adultos:
Nefrolitíase inexplicada
Síndrome de Sjögren com acidose metabólica inexplicada
Crianças:
Má progressão estaturo-ponderal
Osteomalácia
Raquitismo
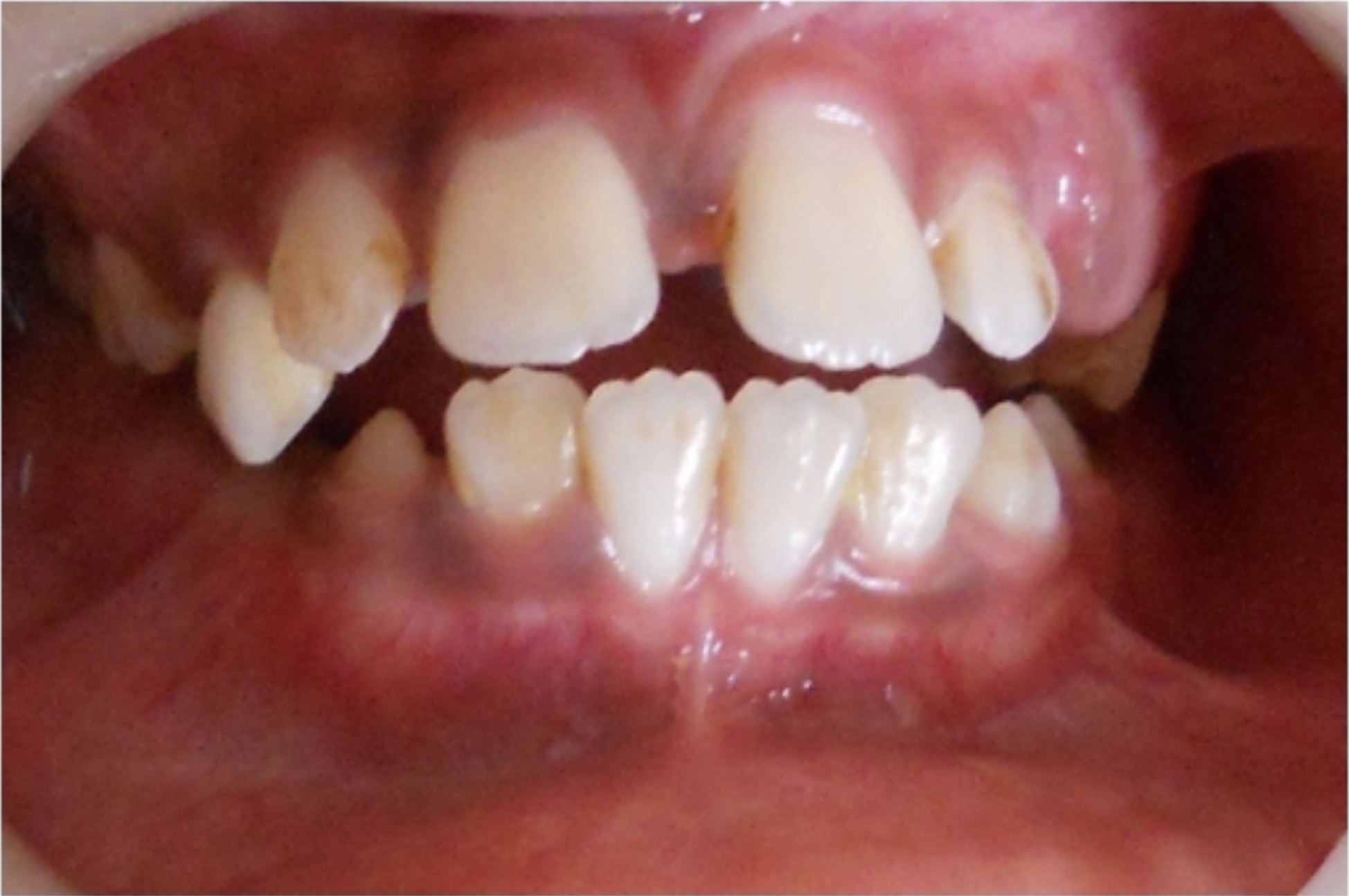
Apresentação dentária de raquitismo, observado na acidose tubular renal distal: O raquitismo pode ser encontrado em indivíduos com acidose tubular renal distal, pois envolve a perda de cálcio na urina.
Imagem: “Showing open bite” por Department of Pedodontics and Preventive Dentistry, Sharad Pawar Dental College, Sawangi (M), Mahartashtra State, Wardha 442102, India. Licença: CC BY 3.0
Acidose tubular renal proximal (tipo 2)
Geralmente ocorre como parte da síndrome de Fanconi, em vez de ocorrer ATR isolada:
A síndrome de Fanconi refere-se à disfunção difusa do túbulo proximal
Alteração da reabsorção de fósforo, ácido úrico, aminoácidos e glicose
Ocorre perda urinária de todas estas substâncias
Adultos:
Indivíduos com mieloma múltiplo e síndrome de Fanconi
Síndrome de Fanconi devido a fármacos
Crianças:
Doença genética que cause síndrome de Fanconi (cistinose, tirosinemia)
Síndrome de Fanconi devido a fármacos
Acidose tubular renal mista (tipo 3)
Observada apenas em áreas onde a consanguinidade é comum:
Norte de África
Arábia Saudita
Observada apenas em crianças:
Defeitos metabólicos muitas vezes incompatíveis com a sobrevivência até à idade reprodutiva
Aqueles que sobrevivem têm poucas probabilidades de se reproduzirem.
Observa-se que as crianças afetadas têm:
Baixa estatura
Doença do “marble brain” → atraso mental
Calcificações cerebrais → surdez e cegueira devido à compressão nervosa
Dismorfismo facial
Osteoporose → fraturas ósseas
Nefrolitíase
Paralisia hipocalémica aguda (associada a insuficiência respiratória diafragmática)
ComaComaComa is defined as a deep state of unarousable unresponsiveness, characterized by a score of 3 points on the GCS. A comatose state can be caused by a multitude of conditions, making the precise epidemiology and prognosis of coma difficult to determine. Coma
Choque
Acidose tubular renal hipercalémica (tipo 4)
Em adultos, geralmente é um achado laboratorial incidental relacionado com:
DiabetesDiabetesDiabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance.Diabetes Mellitus
DRC leve a moderada
Fármacos (AINEs, diuréticos poupadores de K, IECA, ARAs, heparina, trimetoprima)
Em crianças, geralmente é causada por doenças genéticas raras:
Pseudohipoaldosteronismo tipo 2 (síndrome de Gordon)
Hipoaldosteronismo isolado congénito
Diagnóstico
A acidose tubular renal deve ser considerada como diagnóstico diferencial da acidose metabólica sem gap aniónico (NAGMA, pela sigla em inglês).
Passo 1
Uma vez identificada a NAGMA, considerar no diagnóstico diferencial:
ATR
Diarreia
Acidose dilucional (ou seja, soro fisiológico IV excessivo)
DRC (precoce a moderada)
Derivação urinária para o trato GI (ou seja, fístula ureterossigmoide)
Toxicidade do tolueno (cola “huffing”)
Passo 2
Verificar o gap osmolal urinário (UOG, pela sigla em inglês) e/ou o gap aniónico urinário (UAG, pela sigla em inglês).
Diferencia a ATR de outra causa de NAGMA, uma vez que as fórmulas aproximam a excreção urinária de amónio:
As acidoses tubulares renais, geralmente, não cursam com excreção aumentada de amónio.
Outras causas de NAGMA regulam positivamente a excreção de amónio.
O gap aniónico urinário é impreciso em muitas situações clínicas comuns:
Hipovolemia
Aniões urinários não medidos (isto é, cetoacidose)
Disfunção renal
O gap osmolal urinário tem em conta estas condições, mas é impreciso se:
Infeção do trato urinário positiva para ureaseUreaseAn enzyme that catalyzes the conversion of urea and water to carbon dioxide and ammonia.Nocardia/Nocardiosis
Intoxicação por álcool tóxico (e.g., metanol, etilenoglicol)
–20 a –50: NAGMA não causada por ATR (ou seja, diarreia)
Passo 3
Para diferenciar os tipos de ATR, avaliar:
Nível de bicarbonato sérico:
Distal (tipo 1): < 10 mEq/L
Proximal (tipo 2): 14–20 mEq/L
Mista (tipo 3): < 3 mEq/L
Hipercalémica (tipo 4): >15 mEq/L
pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance da urina:
Distal (tipo 1): ≥ 5,5
Proximal (tipo 2): variável
≥ 5,5 se HCO3– sérico > que o limiar de reabsorção do túbulo proximal
No início do curso da doença → ainda não se encontra em estado estacionário
Durante o tratamento com bicarbonato
< 5,5 se HCO3– sérico ≤ que o limiar de reabsorção do túbulo proximal
Não tratado e em estado estacionário
Não ocorre perda de HCO3– urinário
Mista (tipo 3): variável
Hipercalémica (tipo 4): < 5,5
Potássio sérico:
Distal (tipo 1): baixo
Proximal (tipo 2): baixo
Hipercalêmico (tipo 4): alto
Passo 4
Se o diagnóstico ainda não for claro, pode ser realizado um teste de infusão de bicarbonato:
Ajuda a diferenciar a ATR distal (tipo 1) da proximal (tipo 2)
Raramente realizado na prática
O bicarbonato IV é administrado até HCO3– sérico = 18–20 mEq/L.
ATR distal (tipo 1)
O pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance da urina não muda, apesar de ↑ HCO3– sérico
A excreção fracionada de HCO3– é < 3% (nível normal)
ATR proximal (tipo 2)
O pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance da urina aumenta à medida que o HCO3– sérico aumenta.
A excreção fracionada de HCO3– é > 15% (↑ devido à perda de HCO3–).
Tratamento
Princípios gerais na acidose tubular renal distal e proximal
O objetivo da terapêutica é normalizar o bicarbonato sérico.
A base da terapêutica é a reposição de basesBasesUsually a hydroxide of lithium, sodium, potassium, rubidium or cesium, but also the carbonates of these metals, ammonia, and the amines.Acid-Base Balance (ou seja, bicarbonato).
Podem ser usados o bicarbonato ou o citrato oral:
O citrato é convertido em bicarbonato no fígado.
Rácio1:1: 1 mEq de citrato torna-se 1 mEq de bicarbonato.
O citrato é benéfico no tratamento da nefrolitíase.
O potássio sérico deve ser considerado:
As preparações com potássio podem ser preferidas se existir hipocalemia.
As preparações com sódio são usadas de outra forma.
A reposição oral à parte de potássio é frequentemente necessária.
A carga de comprimidos é um grande problema:
A maioria dos comprimidos de bicarbonato e citrato tem doses relativamente baixas de bicarbonato
A dosagem é muitas vezes de vários comprimidos por dose, administrados várias vezes por dia.
Predispõe à baixa adesão terapêutica e a uma má qualidade de vida
Muitas vezes incapazes de normalizar o bicarbonato sérico, independentemente do tratamento
A perda de bicarbonato resulta no agaravamento da hipocalemia:
O HCO3– viaja através do nefrónio ligado ao Na + (NaHCO3).
↑ NaHCO3 → ↑ entrega distal de Na+ às células principais do ducto coletor
↑ Reabsorção de Na+ pelo canal ENaCENaCSodium channels found on salt-reabsorbing epithelial cells that line the distal nephron; the distal colon; salivary ducts; sweat glands; and the lung. They are amiloride-sensitive and play a critical role in the control of sodium balance, blood volume, and blood pressure.Liddle Syndrome → ↑ Secreção de K+ pelo canal ROMK
O tratamento envolve uma abordagem combinada:
Interromper qualquer fármaco/medicação ofensiva, se possível.
Tratar a condição subjacente, se possível.
Suplementar com bicarbonato:
Bicarbonato oral
A hidroclorotiazida pode ser usada para melhorar a carga de comprimidos:
Diurético → contração leve do volume → estimula a reabsorção de Na+ e HCO3– no túbulo proximal
Podem ser usados diuréticos poupadores de K+ para ↓ necessidade de suplementação de K
Pode ser necessária suplementação adicional na síndrome de Fanconi:
Fósforo
Vitamina D
Monitorização laboratorial frequente de HCO3– , K+ e fosfato séricos
Acidose tubular renal mista (tipo 3)
Devido à sua raridade, não há consenso sobre o tratamento da ATR tipo 3.
NaHCO₃ se acidose aguda
Suplementação de potássio
Suplementação de Vitamina D
Litotripsia se cálculos renais
Fixação cirúrgica de fraturas ósseas
Monitorização laboratorial frequente do HCO3– e K+ séricos
Acidose tubular renal hipercalémica (tipo 4)
O tratamento da ATR hipercalémica difere significativamente das outras formas de ATR, principalmente porque a terapêutica com bicarbonato oral não é o tratamento de primeira linha.
Fludrocortisona:
Oral de aldosterona análogo
O efeito colateral da retenção de Na+ pode limitar a sua utilidade
Agrava a hipertensão
Pode levar à sobrecarga de volume (edemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema periférico e pulmonar)
Dieta pobre em K+
Diuréticos da ansa ou tiazídicos (principalmente se não for possível usar fludrocortisona)
A terapêutica oral com bicarbonato geralmente não é usada.
Diagnóstico Diferencial
Mieloma múltiplo: discrasia maligna de células plasmáticas que leva a níveis tóxicos de paraproteína sérica. A apresentação cursa com a síndrome CRAB, que inclui hiperCalcemia, insuficiência Renal, Anemia e lesões/dor óssea (Bone lesions/painPainAn unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons.Pain: Types and Pathways). A ATR proximal (tipo 2) também pode estar presente. O diagnóstico é realizado através da eletroforese de proteínas séricas (SPEP, pela sigla em inglês) com imunofixação (IFE, pela sigla em inglês), que identifica a paraproteína anormal (conhecida como M-spike no SPEP). Exames complementares adicionais incluem a eletroforese de proteínas na urina (UPEP, pela sigla em inglês) e a proporção de cadeias leves livres no soro. O tratamento envolve quimioterapia e transplante de células-tronco hematopoiéticas.
Pseudohipoaldosteronismo tipo 1: doença rara que se apresenta no período neonatal como hiponatremia, hipercalemia e acidose metabólica (ATR hipercalémica tipo 4). O pseudo-hipoaldosteronismo tipo 1 pode ser diagnosticado através da constatação do aumento da atividade da renina plasmática e de um nível elevado de aldosterona. O tratamento inclui suplementação de sódio e terapêutica agressiva com fluidos e eletrólitos.
Cistinose: doença de depósitos lisossómicos autossómica recessiva. É a causa hereditária maisMAISAndrogen Insensitivity Syndrome comum da síndrome de Fanconi em crianças e é causada por uma mutação num geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics que codifica a proteína transportadora cistinosina. A cistina intralisossomal acumula-se em todo o corpo, inclusive nas células do túbulo proximal, durante o 1º ano de vida (resultando em ATR proximal tipo 2 e síndrome de Fanconi). Ocorre dano renal progressivo (incluindo no glomérulo) e a doença renal em estágio final ocorre durante a infância. O tratamento inclui o agente depletor de cistina cisteamina e terapêutica de substituição renal (incluindo transplante renal).
Síndrome de Sjögren: doença autoimune em que tecidos glandulares, como as glândulas salivares e lacrimais, são infiltrados por linfócitos, causando sintomas como olhos secos e boca seca. Há uma ampla gama de manifestações extraglandulares da síndrome de Sjögren, incluindo ATR distal (tipo 1), fenómeno de Raynaud, neuropatia e vasculite cutânea. O diagnóstico é validado pelo exame clínico, estudos serológicos e biópsia das glândulas salivares. É necessária uma abordagem multidisciplinar para tratar os indivíduos afetados e o tratamento visa o alívio dos sintomas.
Lúpus eritematoso sistémico (LES): doença autoimune crónica que causa deposição de imunocomplexos nos órgãos, resultando numa ampla gama de possíveis manifestações sistémicas. As características clínicas típicas incluem erupção cutânea malar, artrite não destrutiva, nefrite lúpica, serosite, citopenias, doença tromboembólica, convulsões e/ou psicose. A ATR hipercalémica (tipo 4), também pode estar presente. O diagnóstico é realizado através do preenchimento de critérios clínicos, que incluem anticorpos antinucleares, anticorpos específicos para LES e achados clínicos específicos. As opções de tratamento incluem hidroxicloroquina e fármacos imunossupressores (e.g., prednisona, metotrexato, micofenolato mofetil).
Tirosinemia: alteração genética autossómica recessiva causada por um defeito na quebra do aminoácido tirosina, resultando em défice de crescimento durante o 1º mês de vida. Outras manifestações clínicas incluem ATR proximal (tipo 2), síndrome de Fanconi, fezes sanguinolentas, odor a repolho, icterícia, hepatomegalia e vómitos. Sem tratamento, desenvolvem-se cirrose e carcinoma hepatocelular. O tratamento inclui nitisinona e uma dieta pobre em tirosina (ou seja, pobre em proteínas).
Doença de Wilson: também conhecida como degeneração hepatolenticular. A doença de Wilson é um distúrbio autossómico recessivo associado a mutações no geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of GeneticsATP7B, que resultam na acumulação de cobre no fígado, cérebro e córnea. A apresentação cursa com sintomas hepáticos, neurológicos e psiquiátricos. A ATR distal (tipo 1), também pode estar presente. O diagnóstico é estabelecido pela deteção de depósitos de cobre na córnea (anéis de Kayser-Fleischer), ceruloplasmina plasmática baixa e/ou níveis elevados de cobre na urina. O tratamento baseia-se no uso de agentes quelantes de cobre, tais como a penicilamina.
Referências
Bello, C., Duarte, J. S., & Vasconcelos, C. (2017). Diabetes mellitus and hyperkalemic renal tubular acidosis: case reports and literature review. J Bras Nefrol 39, pp. 481–485. https://doi.org/10.5935/0101-2800.20170086
Berend, K. (2017). Review of the diagnostic evaluation of normal anion gap metabolic acidosis. Kidney Dis (Basel) 3, pp. 149–159.
Dobbin, S., Petrie, J. R., Lean, M., & McKay, G. A. (2017). Fludrocortisone therapy for persistent hyperkalaemia. Diabetic medicine : a journal of the British Diabetic Association, 34(7), pp. 1005–1008. https://doi.org/10.1111/dme.13359
Karet, F. E. (2009). Mechanisms in hyperkalemic renal tubular acidosis. Journal of the American Society of Nephrology (JASN), 20(2), pp. 251–254. https://doi.org/10.1681/ASN.2008020166
Koratala, A., & Ruchi, R. (2017). Hypokalemia: A potentially life-threatening complication of tenofovir therapy. SAGE open medical case reports, 5. https://doi.org/10.1177/2050313X17741010
Marangella, M. (2017). Impiego del citrato nel paziente con Nefrolitiasi [Use of citrate in patients with nephrolithiasis]. Giornale italiano di nefrologia: organo ufficiale della Societa italiana di nefrologia, 34(4), pp. 51–60.
Goswami, R. P., Mondal, S., Karmakar, P. S., Ghosh, A. (2012). Type 3 renal tubular acidosis. Indian Journal of Nephrology, 22(6), 466–468. doi: 10.4103/0971-4065.106058. PMID: 23439805; PMCID: PMC3573491. Retrieved October 8, 2022, from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3573491/
Alsharidi, A., Al-Hamed, M., Alsuwaida, A. (2016). Carbonic anhydrase II deficiency: report of a novel mutation. CEN Case Reports 5(1), 108, 112. doi: 10.1007/s13730-015-0205-y. PMID: 28509178; PMCID: PMC5411668. Retrieved October 8, 2022, from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5411668/
¡Crea tu cuenta gratis o inicia una sesión para seguir leyendo!
A Lecturio Medical complementa o teu estudo através de métodos de ensino baseados em evidência, vídeos de palestras, perguntas e muito mais – tudo combinado num só lugar e fácil de usar.
User Reviews
Details
×
Obtenha Premium para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características
Obtenha Premium para ver todos os vídeos
Verifique agora o seu e-mail para obter um teste gratuito.
Crie uma conta gratuita para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características - incluindo o Qbank de Lecturio com perguntas actualizadas ao estilo do board-.