Las anomalías de la córnea son condiciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las que la estructura o función de la córnea se veVEVentilation: Mechanics of Breathing afectada debido a diversas patologías congénitas o adquiridas. Las anomalías corneales se clasifican enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función de las anomalías de tamaño, claridad, anomalías ectásicas, distrofias corneales y condiciones adquiridas. LosLOSNeisseria cambios patológicos pueden dar lugar a la opacidad o nubosidad de la córnea y, por lo tanto, reducir la agudeza visual. Las anomalías de la córnea suelen diagnosticarse a partir de losLOSNeisseria hallazgos clínicos. El tratamiento incluye la corrección de losLOSNeisseria errores refractivos y el tratamiento de las condiciones subyacentes.
La curvatura de la córnea es > alALAmyloidosis resto del globo.
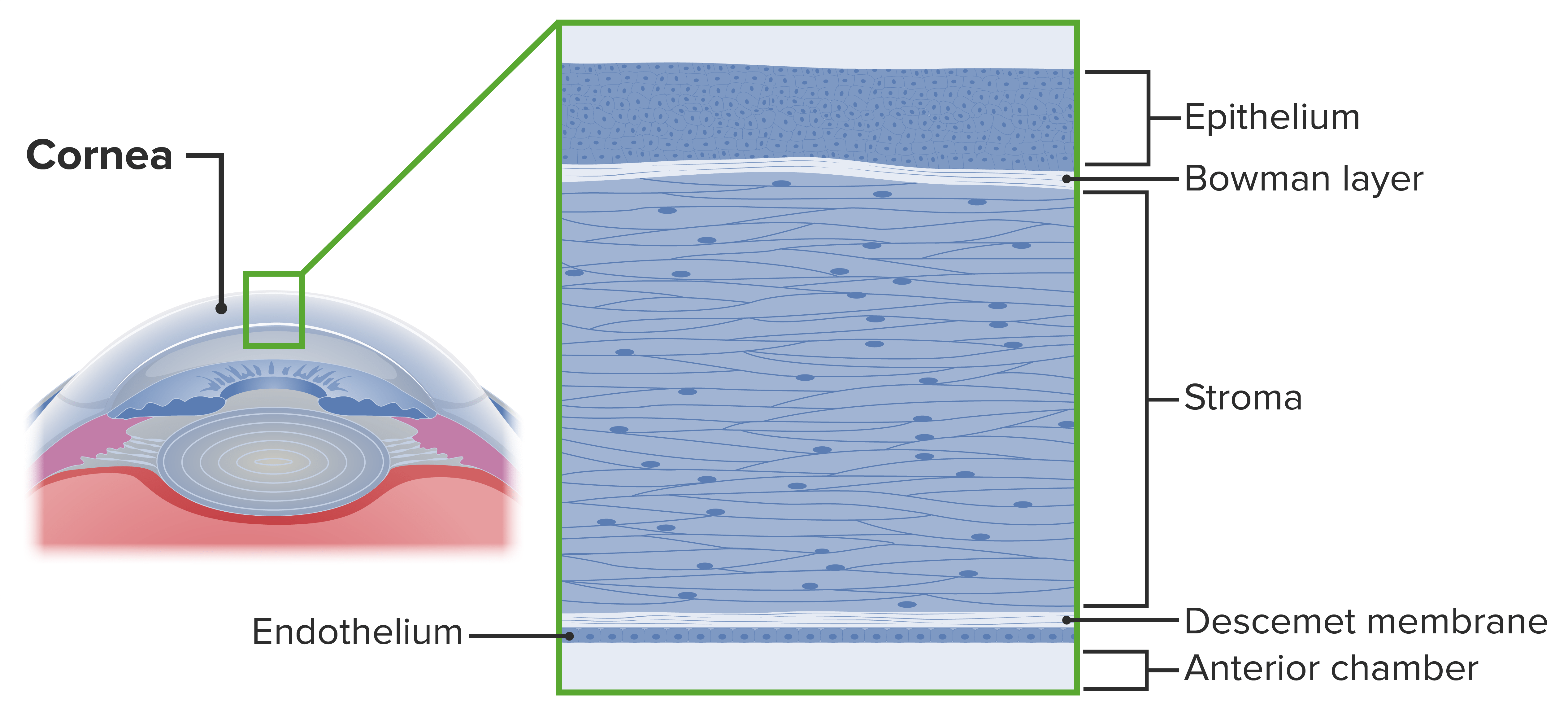
5 capas histológicas:
Epitelio
Membrana de Bowman
Estroma
Membrana de Descemet
Endotelio
Clasificación de las anomalías de la córnea
Criptoftalmos
Anomalías de tamaño:
Microcórnea
Megalocórnea
Anomalías ectásicas:
Queratocono
Queratoglobo
Anomalías de la claridad:
Anomalía de Peters
Esclerocórnea
Distrofias corneales:
Distrofia corneal estromal congénita
Distrofia corneal endotelial congénita
Condiciones adquiridas:
Queloides corneales
Queratopatía enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum banda
Capas de la córnea
Imagen por Lecturio.
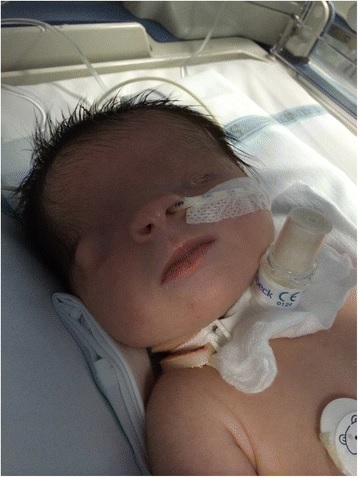
Criptoftalmos
El criptoftalmos es una anomalía congénita autosómica recesiva asociada a anomalías sistémicas y se caracteriza por una continuidad ininterrumpida de la piel que se extiende desde la frente hasta la región malar.
Epidemiología
La incidencia del criptoftalmos es desconocida.
Puede ocurrir de forma aislada
También se asocia con el síndrome de Fraser, un trastorno de malformación autosómico recesivo que se presenta con:
Criptoftalmos
Sindactilia
Anomalías de las vías respiratorias y urogenitales
Bilateral > unilateral
Presentación clínica
Tres formas:
Completa = falla total de la formación del párpado
Tipo más común
LosLOSNeisseria párpados son sustituidos por una capa de piel que se extiende desde la frente hasta la región malar.
Ausencia o escaso desarrollo de la ceja
La córnea está ausente o fusionada con la piel.
Parcial = resultado del desarrollo anormal del pliegue del párpado
Comprende aproximadamente el 20% de losLOSNeisseria casos
LosLOSNeisseria párpados rudimentarios están presentes con un pequeño saco conjuntival situado lateralmente.
El globo ocular es pequeño y está casi completamente cubierto por la piel.
Abortiva = La parte del pliegue del párpado derivada del proceso frontonasal no se desarrolla, mientras que la parte derivada del proceso maxilar se desarrolla con normalidad.
El párpado superior anormal cubre y se adhiere hasta el 75% de la córnea superior.
No hay punctum ni fórnix conjuntival superior
Párpado inferior normal
Tratamiento
El tratamiento está dirigido a la reconstrucción quirúrgica de losLOSNeisseria párpados para permitir el desarrollo visual. No existe un enfoque estándar, y las cirugías difieren según la gravedad de las anomalías y el desarrollo de la órbita.
Criptoftalmos bilateral
Imagen: “Bilateral cryptophthalmos with microphthalmos into left ocular globe and abnormal right ocular globe in a female infant with Fraser syndrome” por De Bernardo et al. Licencia: CC BY 4.0
Anomalías del Tamaño Corneal
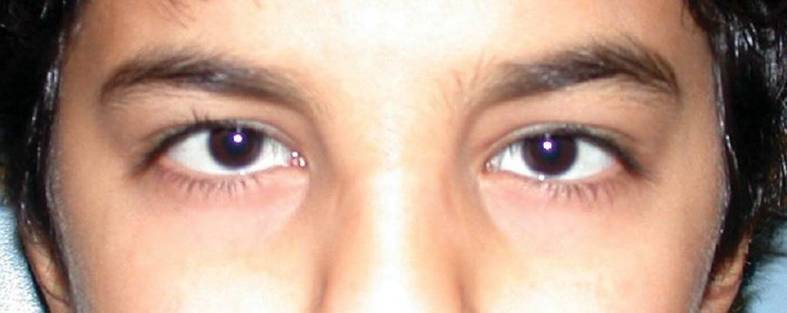
Microcórnea
El diámetro horizontal normal de la córnea alALAmyloidosis nacer es de 10 mm. El tamaño adulto de unos 11,7 mm se alcanza normalmente a losLOSNeisseria 2 años de edad.
Descripción general:
La microcórnea se caracteriza por un diámetro corneal < 10 mm.
Puede ser unilateral o bilateral
Puede ocurrir como una anomalía independiente
Puede estar asociado a:
Microftalmos: globo anormalmente pequeño
Nanoftalmos: ojo anormalmente pequeño con un cristalino de tamaño normal
Presentación clínica:
Suele asociarse a la miopía y a la microftalmos
Puede presentarse como microcórnea aislada o microftalmos anterior relativo
LosLOSNeisseria individuos suelen desarrollar el glaucomaGlaucomaGlaucoma is an optic neuropathy characterized by typical visual field defects and optic nerve atrophy seen as optic disc cupping on examination. The acute form of glaucoma is a medical emergency. Glaucoma is often, but not always, caused by increased intraocular pressure (IOP). GlaucomaenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la 4ta década de la vida.
Niño con microcórnea (diámetros < 10 mm)
Imagen: “At 12 years of age, the child had horizontal corneal diameters of 10 mm but otherwise unremarkable anterior segments by slit-lamp examination” por Hazin R et al. Licencia: CC BY 2.0
La megalocórnea es un agrandamiento bilateral y no progresivo de la córnea que se caracteriza por un diámetro horizontal de la córnea > 12 mm alALAmyloidosis nacer y 13 mm a losLOSNeisseria 2 años de edad.
Descripción general:
Por lo general, es una córnea clara con un grosor y una visión normales
Se hereda como un rasgo recesivo ligado alALAmyloidosis cromosoma X
Asociaciones sistémicas:
Síndrome de Marfan
Síndrome de Ehlers-Danlos
Síndrome de Down
Fisiopatología:
Durante el desarrollo de la córnea, un defecto enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la formación de la copa óptica permite que esta crezca enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum exceso.
Se considera un crecimiento excesivo primario de la córnea
La densidad de las células endoteliales es normal.
Presentación clínica:
Se presenta después de losLOSNeisseria 12 meses de edad, cuando la córnea se haHAHemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis).Hemolytic Anemia desarrollado completamente
Córneas simétricas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum forma de cúpula > 13 mm de diámetro
La reducción de la agudeza visual es poco frecuente.
El astigmatismo es común.
Megalocórnea en un niño con un diámetro de córnea > 14 mm
Imagen: “Megalocornea in a child with a corneal diameter greater than 14 mm” por Khan AO et al. Licencia: CC BY 3.0, recortada por Lecturio.
Anomalías Ectásicas de la Córnea
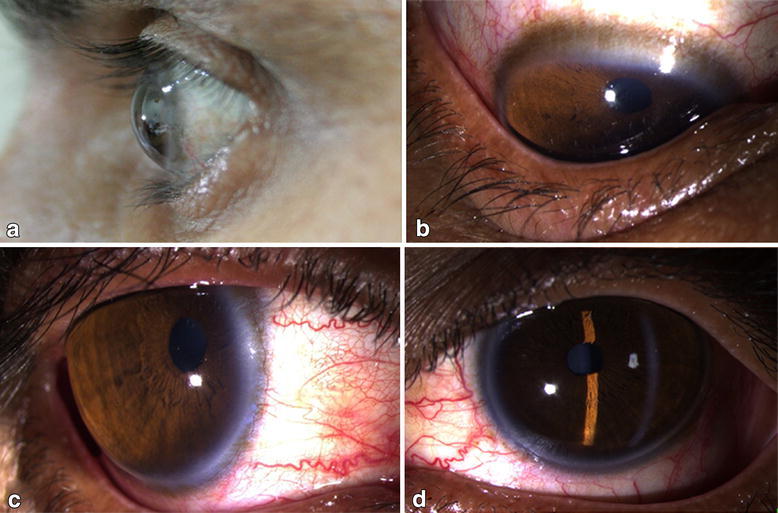
Queratocono
El queratocono se refiere alALAmyloidosis adelgazamiento cerca del centro de la córnea, lo que da lugar a la inclinación de la córnea y a su forma cónica.
Descripción general:
No inflamatorio
Trastorno progresivo
Causado por una debilidad congénita de la córnea
Puede ser secundario a una queratoconjuntivitis viral o a un traumatismo
Presentación clínica:
Se presenta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la pubertad o enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la edad adulta temprana
El deterioro de la visión progresa hasta la 4ta década.
Usualmente bilateral, pero suele ser más grave enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un ojo
Se presentan dificultades para corregir la visión por miopía y astigmatismo
Hallazgos con enfermedad avanzada:
Signo de Munson: hendidura enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum forma de V del párpado inferior cuando la persona mira hacia abajo (ver imagen)
Estrías de Vogt: líneas de tensión estromal profundas que son patognomónicas del queratocono
Anillo de Fleischer: depósito de hemosiderina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la base del cono
Hidropesía aguda: Pueden producirse roturas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la membrana de Descemet que hacen que el estroma se vuelva repentinamente edematoso y opaco.
Puede presentarse con fotofobia y una caída repentina y dolorosa de la agudeza visual
LosLOSNeisseria tratamientos incluyen parches de presión y lentes de contacto como vendaje.
Se utilizan hiperosmóticos, ciclopléjicos y esteroides tópicos para disminuir el edemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema, el dolorDolorInflammation y la inflamación.
Diagnóstico:
Examen con lámpara de hendidura: La córnea puede parecer normal.
Queratometría: puede mostrar distorsión
Topografía corneal: puede ayudar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el diagnóstico y enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la evaluación de la progresión de la enfermedad
Tratamiento:
Reticulación (crosslinking) del colágeno corneal: Procedimiento que utiliza gotas de riboflavina, luz UV y un fotosensibilizador para reforzar losLOSNeisseria enlaces enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la córnea y medir la progresión.
Anteojos: para corregir el astigmatismo miópico mientras el paciente pueda utilizarlos
Lentes de contacto:
LosLOSNeisseria lentes rígidos permeables alALAmyloidosis gas neutralizan la irregularidad de la córnea, pero pueden llegar a ser intolerables.
Se pueden utilizar lentes esclerales que se asientan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la esclera sin tocar la córnea.
Intervenciones quirúrgicas:
Queratoplastia: necesaria enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum 10%–15% de losLOSNeisseria pacientes
Segmentos de anillos corneales intraestromales
El signo de Munson en un paciente con queratocono
Imagen: “Münson’s sign in a keratoconus patient which appears as bulging of the lower lid during downgaze” por BioMed Research International/Mariam Lotfy Khaled et al. Licencia: CC BY 4.0
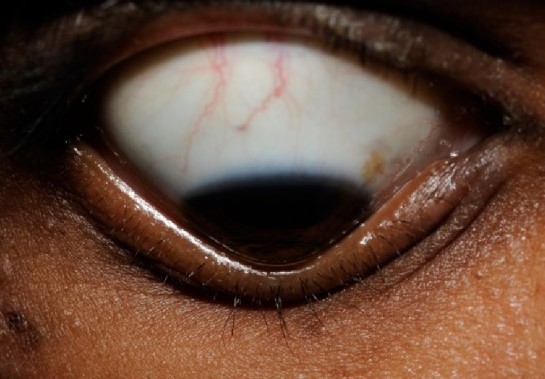
Queratoglobo
El queratoglobo es una protrusión hemisférica de toda la córnea con un adelgazamiento corneal de limbo a limbo.
Descripción general:
Si es congénita, la afección es bilateral.
También se han visto formas adquiridas debido a:
Queratoconjuntivitis
Blefaritis marginal crónica
Inflamación orbital idiopática
De origen no inflamatorio
De naturaleza progresiva
Estrechamente relacionado con el síndrome de Ehlers-Danlos tipo VI
Suele asociarse a defectos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la síntesis o degradación del colágeno
Presentación clínica:
Córneas claras, a menos que losLOSNeisseria pacientes tengan episodios de hidropesía y cicatrización
Miopía alta con astigmatismo irregular
Perforaciones de la córnea por adelgazamiento extremo
Tratamiento:
Corrección refractiva de la miopía alta
Las gafas de protección reducen la incidencia de las perforaciones corneales.
Adaptación de lentes esclerales personalizados
a: Adelgazamiento difuso de la córnea con protrusión del globo hacia el exterior b: Hendidura del párpado inferior c y d: Limbo estirado con cámara anterior clara
Imagen: “Clinical picture of the patient showing diffuse thinning of cornea with outward globular protrusion” por Gupta N et al. Licencia: CC BY 4.0
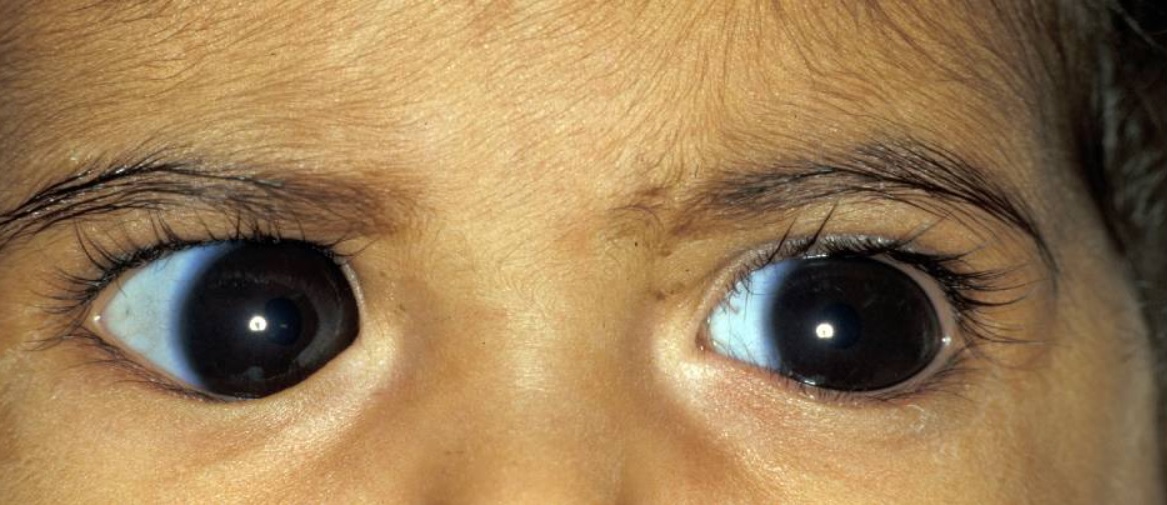
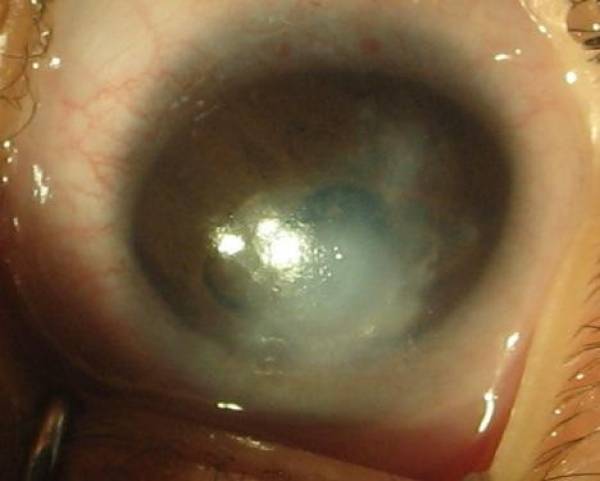
La anomalía de Peters es una forma congénita de disgenesia del segmento anterior enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la que la divisón anormal de la cámara anterior da lugar a un defecto central enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el endotelio corneal y provoca un leucoma (placa blanquecina).
Descripción general:
Múltiples loci genéticos:
PAX6
PITX2
FOXC1
CYP1B1
Puede ocurrir de forma esporádica, pero son comunes las herencias dominantes y recesivas
Las asociaciones sistémicas incluyen:
Trisomía 13
Trisomía 15
Deleción parcial del brazo cromosómico 11q
La incidencia es desconocida: parte de las opacidades corneales congénitas se observan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum 3–6 individuos por cada 100 000
Patogénesis: debido a una alteración enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la migración o separación de la cresta neural que se produce enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la 7ma semana de gestación
Presentación clínica y diagnóstico:
Siempre se presenta una opacidad corneal central, paracentral o completa.
Usualmente, se observan hebras de iris a través de la cámara anterior hasta la superficie posterior de la córnea.
La anomalía de Peters tipo 1:
Se presenta con opacidad corneal central
Se presenta con adherencias iridocorneales.
Anomalía de Peters tipo 2:
Asociado a una opacificación corneal más densa
Presenta adhesión queratolenticular
Diagnóstico: basado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria hallazgos clínicos y confirmado mediante ultrasonido
Tratamiento:
Control regularRegularInsulin del glaucomaGlaucomaGlaucoma is an optic neuropathy characterized by typical visual field defects and optic nerve atrophy seen as optic disc cupping on examination. The acute form of glaucoma is a medical emergency. Glaucoma is often, but not always, caused by increased intraocular pressure (IOP). Glaucoma
Cirugía: queratoplastia penetrante de espesor total más lensectomía enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes con cataratas
Opacidad corneal en un paciente con anomalía de Peters
Imagen: “Photograph of left eye” por Tuli N et al. Licencia: CC BY 2.0
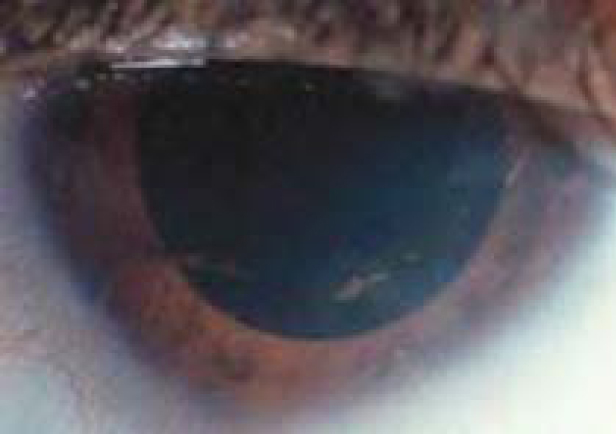
Esclerocórnea
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la esclerocórnea, losLOSNeisseria límites de la córnea y la esclerótica son indistintos, lo que resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum una anomalía congénita de claridad. Se observan fibrillas de colágeno dispuestas de forma irregular.
Descripción general:
Suelen presentarse anomalías oculares adicionales, como cataratas o colobomas.
No progresivo
No inflamatorio
Puede afectar a toda la córnea o limitarse a una parte de la misma:
Esclerocórnea periférica: La zona afectada está vascularizada con vasos esclerales superficiales.
Esclerocórnea total: Toda la córnea es opaca y está vascularizada.
Etiología:
Forma autosómica dominante o autosómica recesiva (mucho más grave)
Suele ocurrir de forma bilateral
Fisiopatología:
El desarrollo de la córnea se produce durante la 7ma y la 8va semana de la gestación.
El hecho de que las células mesenquimales no se diferencien enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum células corneales y esclerales permite que la curvatura de la córnea supere la de la esclera.
Presentación clínica:
Esclerocórnea parcial: presencia de un borde corneal periférico, blanco, vascularizado, de 1–2 mm, que se mezcla con la esclerótica. La córnea central es generalmente normal.
Esclerocórnea total:
Toda la córnea está afectada.
El centro es más claro que la periferia.
Tratamiento:
Por lo general, no se requiere de ningún tratamiento.
LosLOSNeisseria errores de refracción se corrigen si están presentes.
Se pueden utilizar lágrimas artificiales.
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la esclerocórnea generalizada, debe considerarse la posibilidad de realizar una queratoplastia temprana para proporcionar y/o preservar la visión.
Cambios esclerales periféricos observados en un paciente con esclerocórnea
Imagen: “Sclerocornea” por Mataftsi A et al. Licencia: CC BY 3.0
Distrofias Corneales
Distrofia corneal estromal congénita
Trastorno corneal hereditario no inflamatorio que suele no estar asociado a ninguna otra afección ocular o sistémica
Descripción general:
Suele afectar a la zona central de la córnea
Se desarrollan áreas discretas de opacificación enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las capas superficiales del estroma.
Etiología:
Causado por mutaciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen DCN
Autosómico dominante
Clasificación:
Distrofia reticular, tipos I y II
Distrofia granular, tipos I y II
Distrofia macular
Distrofia de Reis-Bucklers
Fisiopatología:
LosLOSNeisseria síntomas clínicos surgen debido a la atrofia y degeneración de las células epiteliales basales.
Se produce un aumento del número y la densidad hasta que la membrana de Bowman se erosiona y el epitelio se descama.
Las distrofias corneales pueden presentarse como formas granulares, reticulares o maculares.
La superficie corneal es normal o ligeramente irregular; se observan pequeñas opacidades enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum todo el estroma de la córnea, dando un aspecto nuboso.
Presentación clínica y diagnóstico:
Reducción de la visión debido a losLOSNeisseria depósitos
El estrabismo es frecuente.
Examen con lámpara de hendidura: La córnea anterior muestra opacidades escamosas o enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum forma de pluma.
Tratamiento:
Anteojos o lentes de contacto para corregir errores de refracción
Parche o corrección quirúrgica del estrabismo
Queratoplastia
Distrofia corneal endotelial congénita
La distrofia endotelial hereditaria congénita es una distrofia corneal caracterizada por la opacidad bilateral difusa de las córneas.
Etiología y epidemiología:
Autosómico recesivo
Mutación enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cromosoma 20p13
Gen afectado: SLC4A11
Incidencia: 3 por cada 100 000 nacimientos
Es más común enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum hijos de matrimonios consanguíneos.
Clasificación:
Distrofia corneal endotelial congénita
Distrofia corneal polimorfa posterior
Fisiopatología:
El gen SLC4A11 codifica una proteína transmembrana, que actúa como bomba entre el endotelio y el estroma corneal.
La disfunción de esta proteína conduce alALAmyloidosis depósito de material enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las capas de la córnea y a la opacidad corneal.
Presentación clínica y diagnóstico:
Antecedentes de opacidad congénita o perinatal de las córneas bilateralmente
Hallazgos del examen físico:
Visión reducida
Ambliopía
Nistagmo
Tratamiento:
La cirugía es el pilar del tratamiento.
Se utiliza la queratoplastia penetrante o la queratoplastia endotelial automatizada de Descemet.
Afecciones Corneales Adquiridas
Queloides corneales
LosLOSNeisseria queloides corneales son lesiones benignas, poco frecuentes y de color blanco-grisáceo, que se producen por la proliferación anormal de tejido fibroso. La acumulación de colágeno y de diversas glicoproteínas da lugar a la hiperplasia del epitelio y a la alteración de la membrana de Bowman.
Descripción general:
Congénito: LosLOSNeisseria casos bilaterales de queloides corneales están asociados alALAmyloidosis síndrome de Lowe y alALAmyloidosis síndrome de Rubinstein-Taybi.
Adquirido: puede desarrollarse luego de un traumatismo ocular o una infección
Se haHAHemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis).Hemolytic Anemia registrado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum edades comprendidas entre losLOSNeisseria 2 meses y losLOSNeisseria 72 años, pero suele ser poco frecuente
La mayoría de losLOSNeisseria queloides corneales se producen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las primeras 3 décadas de vida.
Patogénesis: El crecimiento excesivo del estroma corneal durante el proceso de cicatrización da lugar a la transformación de losLOSNeisseria queratocitos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum fibroblastos y miofibroblastos, lo que conduce a la formación de queloides.
Presentación clínica:
La mayoría de losLOSNeisseria pacientes presentan una lesión corneal de crecimiento lento que parece estar elevada de la superficie corneal.
Aspecto blanco-grisáceo
Pérdida de visión indolora y progresiva
Dificultad para cerrar losLOSNeisseria párpados enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el caso de queloides grandes
El diagnóstico se realiza mediante losLOSNeisseria exámenes de agudeza visual y con lámpara de hendidura.
Tratamiento:
No se necesita tratamiento cuando es asintomático
Monitoreo estrecho:
Tamaño del queloide
Agudeza visual
Se considera la cirugía cuando el queloide provoca una disminución de la agudeza visual:
Escisión local
Queratectomía superficial lamelar o fototerapéutica
Queratoplastia
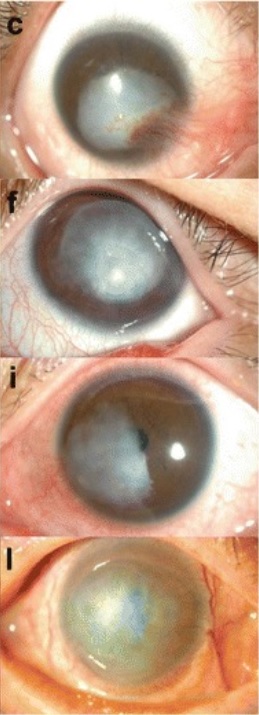
Los queloides corneales aparecen como opacidades que surgen de la superficie epicorneal.
Imagen: “Slit-lamp microscopic photographs of corneal keloids” por BMC Ophthalmology/Hyo Kyung Lee et al. Licencia: CC BY 4.0, recortada por Lecturio.
Queratopatía enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum banda
Cambio degenerativo asociado alALAmyloidosis depósito de sales de calcio enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la membrana de Bowman y enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las láminas del estroma anterior del epitelio corneal
Descripción general:
El depósito puede deberse a diversos factores:
Precipitación de lágrimas
Cambio de pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance
Compromiso de la función endotelial
EdemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema corneal
El resto de la córnea es clara.
Se presenta como una opacidad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum forma de banda enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la zona interpalpebral, con un intervalo claro entre losLOSNeisseria extremos de la banda y el limbo
Causada por una inflamación ocular crónica debida a:
Uveítis
Queratouveítis herpética crónica
Ptisis bulbi
Queratoconjuntivitis seca
Artritis idiopática juvenil
Queratitis por herpes zóster
Puede asociarse a trastornos sistémicos:
Hiperparatiroidismo
Toxicidad por vitamina D
ERC
Hipofosfatasia
SarcoidosisSarcoidosisSarcoidosis is a multisystem inflammatory disease that causes noncaseating granulomas. The exact etiology is unknown. Sarcoidosis usually affects the lungs and thoracic lymph nodes, but it can also affect almost every system in the body, including the skin, heart, and eyes, most commonly. Sarcoidosis
Presentación clínica:
La disminución de la agudeza visual es directamente proporcional a la cantidad de depósitos de calcio enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la córnea.
Sensación de cuerpo extraño
Irritación ocular con enrojecimiento conjuntival
Fotofobia
El depósito de calcio comienza enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la periferia y progresa hacia el interior.
Tratamiento:
Desbridamiento superficial y queratectomía lamelar
Tratar la causa subyacente, si se conoce, para disminuir el depósito de calcio enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la córnea.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.