


A síndrome de Wiskott-Aldrich (SWA), também conhecida como síndrome de eczema-trombocitopenia-imunodeficiência, IMD2 IMD2 Wiskott-aldrich syndrome (WAS), also known as eczema-thrombocytopenia-immunodeficiency syndrome, IMD2, or immunodeficiency 2, is a rare genetic mixed disorder of B- and T-cell deficiency that follows an X-linked recessive inheritance pattern. It is caused by a WAS gene mutation that leads to impaired actin cytoskeleton, phagocytosis and chemotaxis, impaired platelet development, and, in general, a loss of humoral and cellular responses. Wiskott-Aldrich Syndrome ou imunodeficiência 2, é um distúrbio genético misto raro de deficiência de células B e T, que segue um padrão de hereditariedade recessivo ligado ao X. É causada por uma mutação no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics WAS que leva ao comprometimento do citoesqueleto de actina, fagocitose e quimiotaxia, comprometimento do desenvolvimento plaquetário e, em geral, perda das respostas humorais e celulares. A síndrome de Wiskott-Aldrich apresenta-se como uma tríade clássica de eczema Eczema Atopic dermatitis, also known as eczema, is a chronic, relapsing, pruritic, inflammatory skin disease that occurs more frequently in children, although adults can also be affected. The condition is often associated with elevated serum levels of IgE and a personal or family history of atopy. Skin dryness, erythema, oozing, crusting, and lichenification are present. Atopic Dermatitis (Eczema), diátese hemorrágica e infeções oportunistas recorrentes, mas também apresenta alto risco de desenvolvimento de autoimunidade e malignidade. O tratamento inclui transplante de células estaminais hematopoiéticas (TCEH), terapêutica de reposição de imunoglobulinas, antibióticos profiláticos e terapêuticos e esteroides sistémicos. Sem o TCEH, o prognóstico é mau e a expectativa de vida é reduzida.
Last updated: Dec 15, 2025
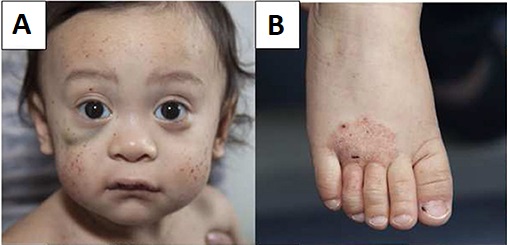
Doente jovem do sexo masculino com síndrome de Wiskott-Aldrich que apresenta múltiplas petéquias faciais, hematoma sob o olho direito (A) e eczema do pé (B)
Imagem: “Wiskott–Aldrich syndrome” por Michael H. Albert and Alexandra F. Freeman, on behalf of the Inborn Errors Working Party (IEWP) of the European Society for Blood and Marrow Transplantation (EBMT). Licença: CC BY 4.0As seguintes condições são diagnósticos diferenciais da síndrome de Wiskott-Aldrich:
