


A sequência de Pierre Robin, também conhecida como síndrome de Pierre Robin ou simplesmente sequência de Robin, é uma condição em bebés caracterizada por uma mandíbula menor que o normal, uma língua que se retrai de volta para a garganta e dificuldade em respirar. A etiologia exata da sequência de Pierre Robin é desconhecida, embora alguns fatores contribuintes tenham sido identificados. O desenvolvimento anormal do maxilar inferior durante a gestação, associado a certas mutações genéticas, pode ser o primeiro de uma série de etapas que levam a problemas respiratórios e de alimentação no recém-nascido.
Last updated: Dec 15, 2025
A sequência de Pierre Robin (SPR) é uma tríade de micrognatia congénita (mandíbula inferior pequena), glossoptose (retração da língua para dentro da faringe) e obstrução das vias aéreas.
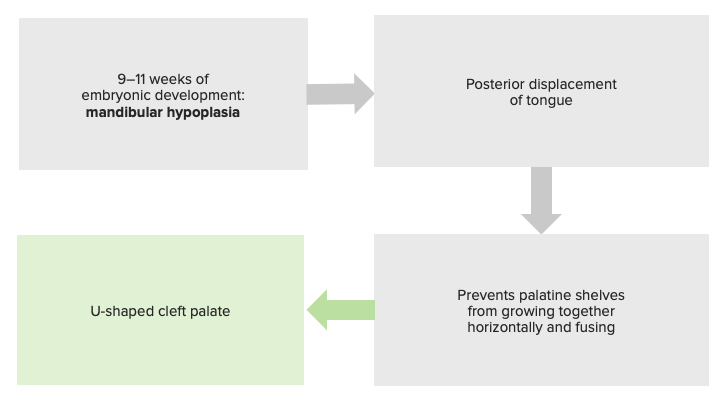
Fisiopatologia da sequência de Pierre Robin, justificando a sua classificação como sequência e não como síndrome.
Imagem por Lecturio.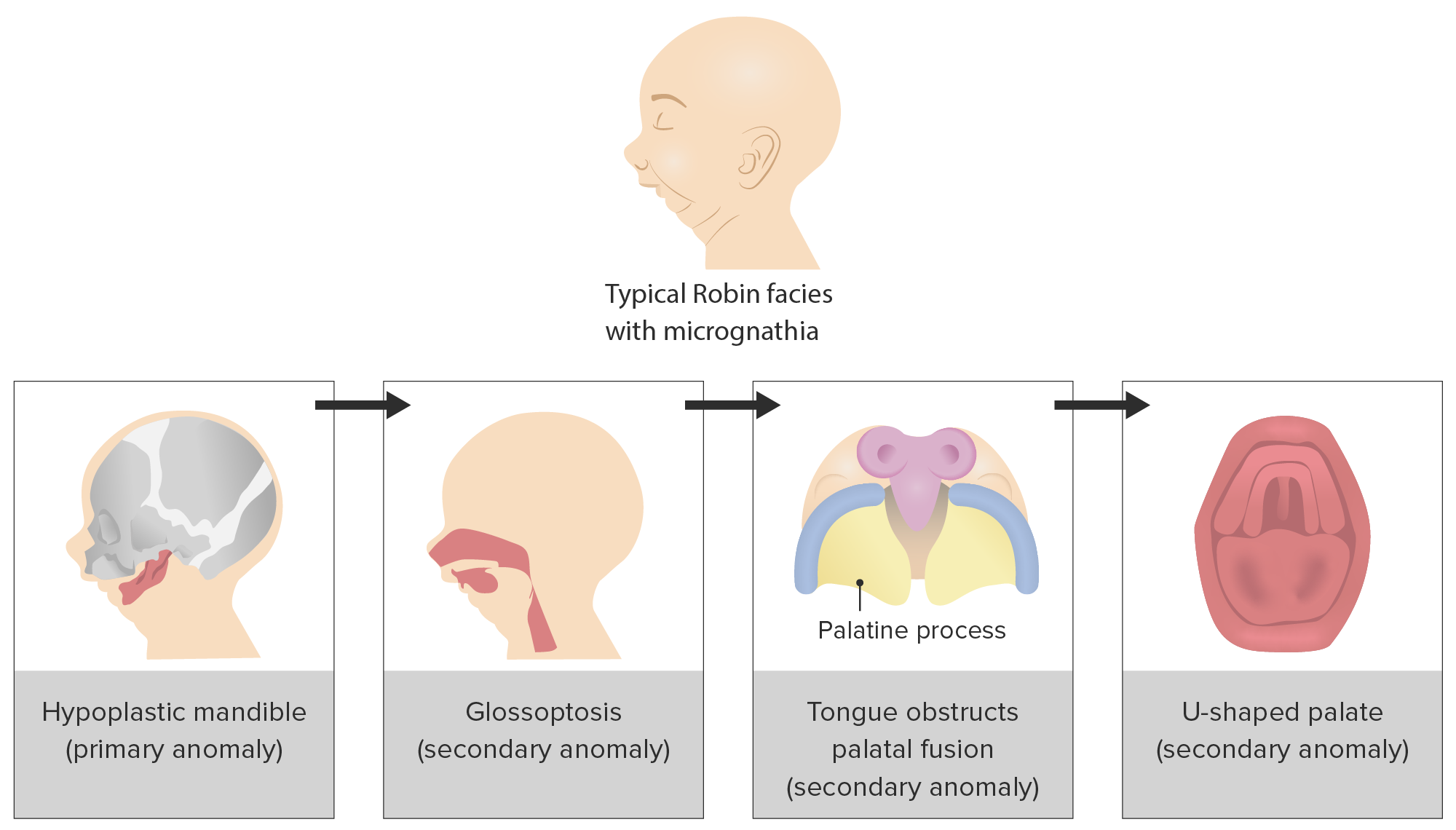
Progressão da tríade de eventos subjacente à apresentação clínica da sequência de Pierre Robin
Imagem por Lecturio.Epidemiologia: 5.15 por 10.000 nados vivos nos Estados Unidos
Etiologia:
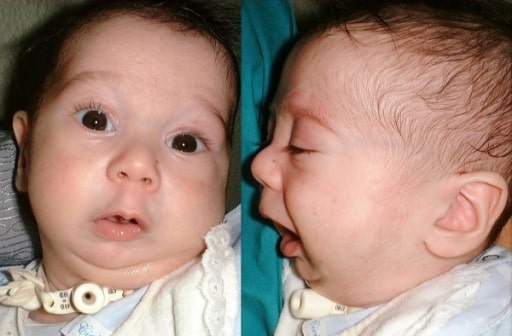
Vista frontal e lateral de uma criança com sequência de Pierre Robin
Imagem: “Preoperative frontal and lateral views of an infant with Pierre Robin sequence” por Maxillo-Facial Surgery Division, Head and Neck Department, University and Hospital of Parma, Parma, Italy. Licença: CC BY 2.0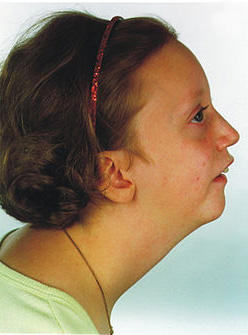
Micrognatia
Imagem: “Pitt-rogers-danks syndrome” por Marie Sogaard et al. Licença: CC BY 2.0, editado por Lecturio.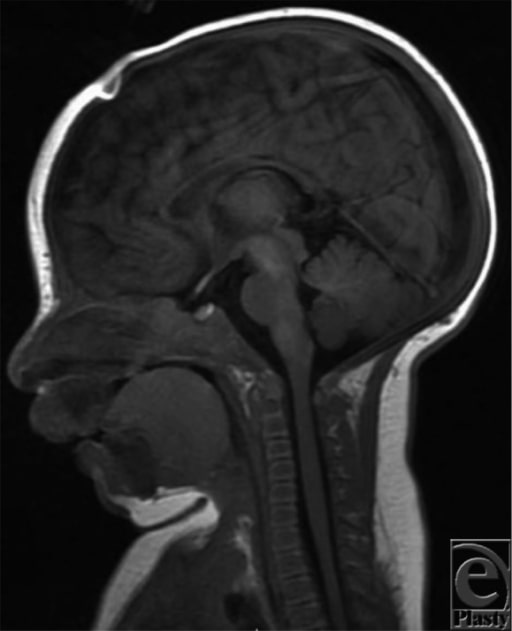
Ressonância magnética (RMN) aos 2 meses de idade indicando hipoplasia mandibular e deslocamento posterior da língua, causando obstrução significativa das vias aéreas faríngeas
Imagem: “Magnetic resonance imaging” por Michigan State University College of Human Medicine, Grand Rapids, Mich. Licença: CC BY 2.0As condições seguintes são diagnósticos diferenciais para a sequência de Pierre Robin isolada ou sindrómica:
