


A doença de Tay-Sachs é uma doença autossómica recessiva de armazenamento lisossómico causado por mutações genéticas no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics da hexosaminidase A Hexosaminidase A A mammalian beta-hexosaminidase isoform that is a heteromeric protein comprised of both hexosaminidase alpha and hexosaminidase beta subunits. Deficiency of hexosaminidase A due to mutations in the gene encoding the hexosaminidase alpha subunit is a case of Tay-sachs disease. Deficiency of hexosaminidase A and hexosaminidase B due to mutations in the gene encoding the hexosaminidase beta subunit is a case of Sandhoff disease. Tay-Sachs Disease(HEXA), levando a neurodegeneração progressiva. Esta doença é altamente prevalecente na população judaica Ashkenazi. Os sintomas clássicos em bebés incluem degeneração rápida das capacidades cognitivas e neuromusculares, cegueira progressiva e uma mancha macular vermelha-cereja no exame físico. O diagnóstico é feito pela medição da atividade da enzima hexosaminidase A Hexosaminidase A A mammalian beta-hexosaminidase isoform that is a heteromeric protein comprised of both hexosaminidase alpha and hexosaminidase beta subunits. Deficiency of hexosaminidase A due to mutations in the gene encoding the hexosaminidase alpha subunit is a case of Tay-sachs disease. Deficiency of hexosaminidase A and hexosaminidase B due to mutations in the gene encoding the hexosaminidase beta subunit is a case of Sandhoff disease. Tay-Sachs Disease e beta-hexosaminidase. Atualmente, não existe cura para esta doença e o tratamento procura melhorar a qualidade de vida do paciente.
Last updated: Apr 23, 2025
A doença de Tay-Sachs é uma doença de armazenamento lisossómico causado por uma deficiência na enzima hexosaminidase A Hexosaminidase A A mammalian beta-hexosaminidase isoform that is a heteromeric protein comprised of both hexosaminidase alpha and hexosaminidase beta subunits. Deficiency of hexosaminidase A due to mutations in the gene encoding the hexosaminidase alpha subunit is a case of Tay-sachs disease. Deficiency of hexosaminidase A and hexosaminidase B due to mutations in the gene encoding the hexosaminidase beta subunit is a case of Sandhoff disease. Tay-Sachs Disease (Hex-A).
A doença de Tay-Sachs é classificada em 3 variantes, com base na idade de início e na atividade enzimática.
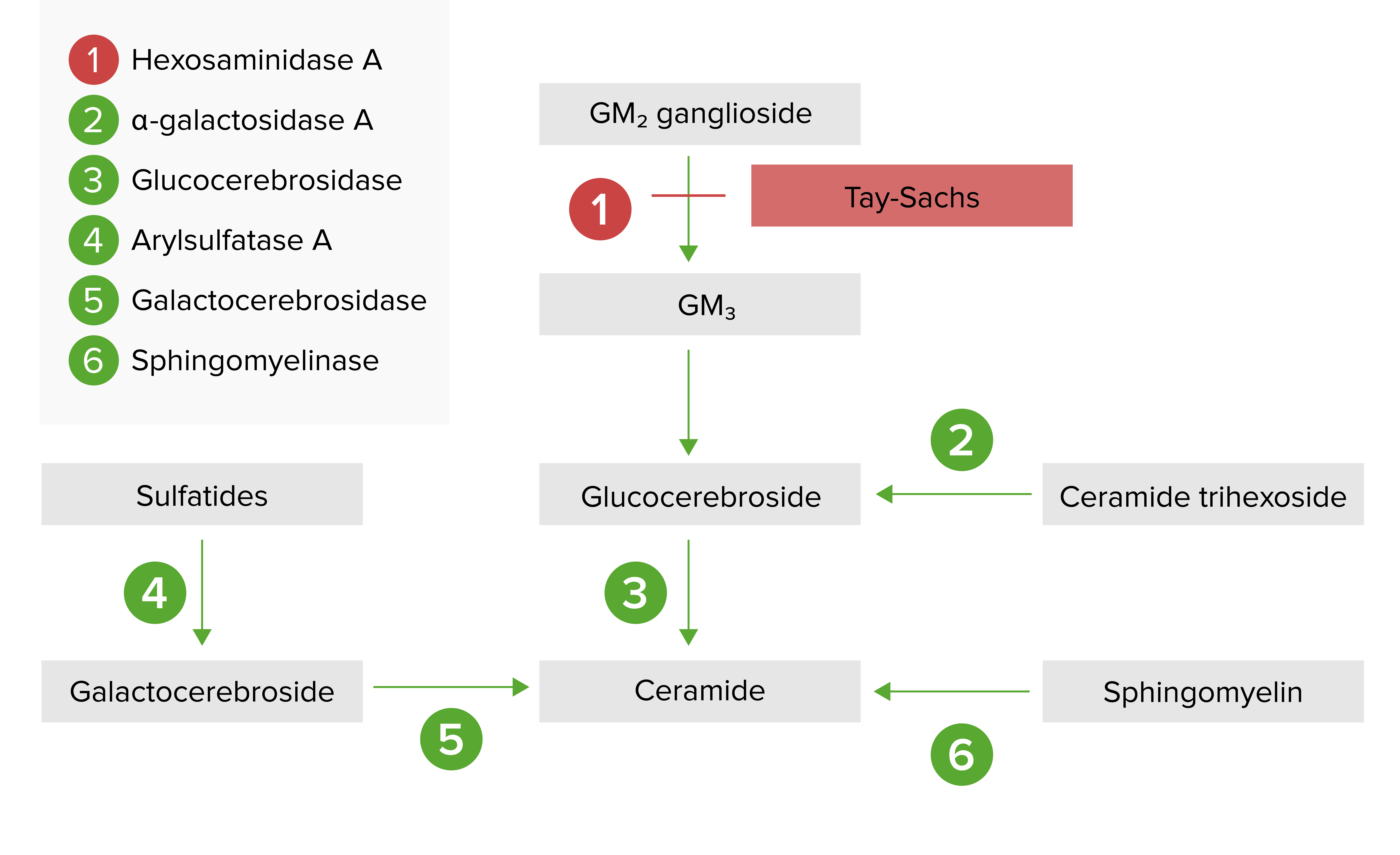
A via de armazenamento lisossomal:
A doença de Tay-Sachs resulta da deficiência de hexosaminidase A, o que resulta num acúmulo do gangliosídio GM2.
A doença de Tay-Sachs é uma doença neurodegenerativa com uma apresentação clínica variável, dependendo da variante.
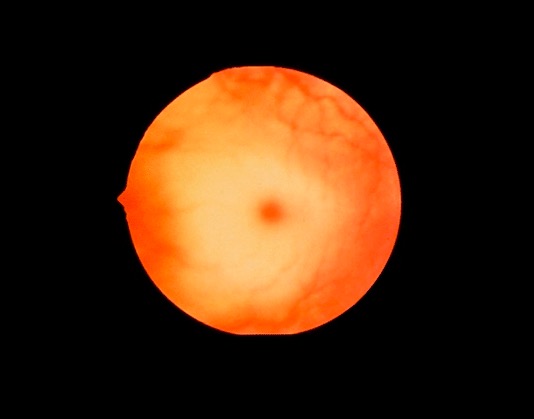
Mancha vermelha-cereja:
A mácula é normalmente vermelha. A palidez desenvolve-se à volta da mácula vermelha devido à acumulação de glicolípidos, dando a aparência de uma mancha vermelha-cereja. A mancha geralmente desenvolve-se até ao 1.º ano de idade na doença de Tay-Sachs, e também pode ser encontrada em outras doenças de depósito lisossómico.
São feitos testes Testes Gonadal Hormones após suspeita clínica. O rastreio pré-natal também é possível através da recolha de amostras de vilosidades coriónicas ou amniocentese.
Não há cura para a doença de Tay-Sachs. O tratamento é de apoio, focado no controlo dos sintomas e na melhoria da qualidade de vida.
