La enfermedad de Von Hippel-Lindau es una condición genética autosómica dominante que resulta de una deleción o mutación en el gen VHL. Los pacientes diagnosticados con la enfermedad de Von Hippel-Lindau tienen tumores y quistes en varias partes de sus cuerpos y pueden presentar hemangioblastomas, carcinoma de células renales, feocromocitomas, tumores del saco endolinfático del oído medio, tumores pancreáticos y cistadenomas papilares del epidídimo o del ligamento ancho. El diagnóstico se realiza mediante pruebas genéticas, pruebas de laboratorio del nitrogeno de urea (BUN, por sus siglas en inglés), pruebas de laboratorio para detectar la presencia de catecolaminas en la sangre o en la orina, examen de fondo de ojo para detectar hemangioblastoma de la retina y TC/RM para detectar cualquier otro tumor. El tratamiento de la enfermedad incluye la extirpación quirúrgica de los tumores.
Last updated: Dec 15, 2025
La enfermedad de Von Hippel-Lindau es un trastorno autosómico dominante caracterizado por hemangioblastomas Hemangioblastomas A benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia. Von Hippel-Lindau Disease de la retina Retina The ten-layered nervous tissue membrane of the eye. It is continuous with the optic nerve and receives images of external objects and transmits visual impulses to the brain. Its outer surface is in contact with the choroid and the inner surface with the vitreous body. The outermost layer is pigmented, whereas the inner nine layers are transparent. Eye: Anatomy y el SNC; quistes que afectan los LOS Neisseria riñones, el páncreas y el epidídimo; carcinoma de células renales; feocromocitomas; y tumores de células de los LOS Neisseria islotes pancreáticos.
La presentación varía según el tamaño y la ubicación del tumor Tumor Inflammation. Los LOS Neisseria antecedentes familiares son una pieza clave de información, ya que la mayoría de los LOS Neisseria casos son hereditarios. El examen físico por lo general no revela gran información, con la excepción de los LOS Neisseria pacientes afectados que presentan anomalías neurológicas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el contexto de los LOS Neisseria hemangioblastomas Hemangioblastomas A benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia. Von Hippel-Lindau Disease.
Se puede recordar el cuadro clínico usando el acrónimo “HIPPEL” ( en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) es:
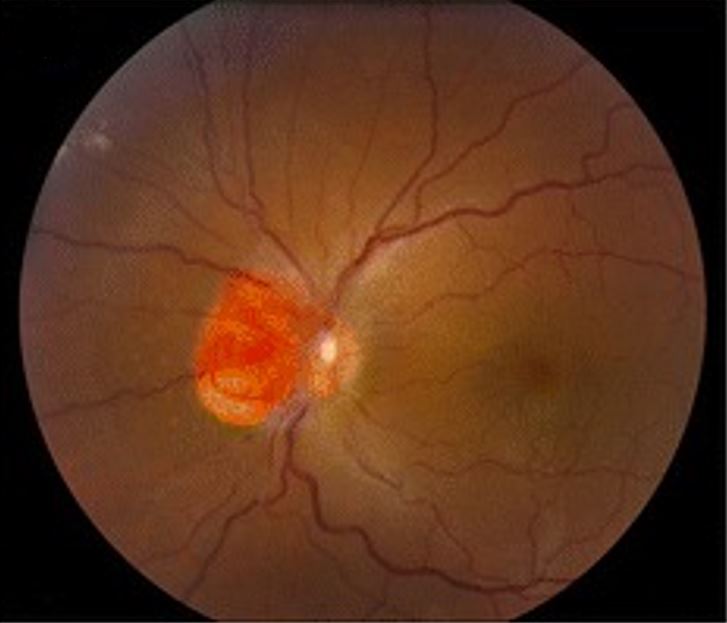
Hemangioblastoma que afecta al nervio óptico, como se observa en la enfermedad de von Hippel-Lindau
Imagen: “Clinical photographs of the eye from the patient with von Hippel-Lindau disease” por Chen S, Chew EY, Chan CC. Licencia: CC BY 4.0, editada por Lecturio.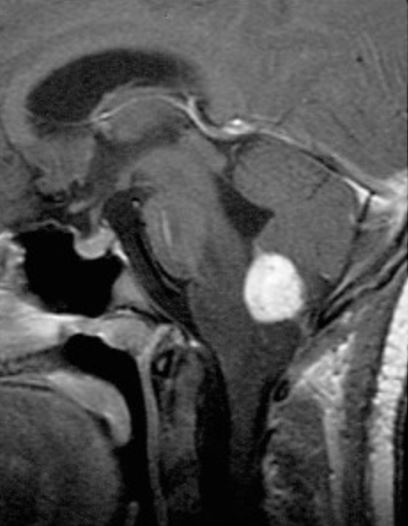
RM sagital, contrastada y ponderada en T1 que muestra un hemangioblastoma medular con realce al contraste
Imagen: “Radiographic images of hemangioblastomas” por Schunemann V, Huntoon K, Lonser RR Licencia: CC BY 4.0, editada por Lecturio.El tratamiento de la enfermedad de Von Hippel-Lindau se adapta según la ubicación y el tamaño de las lesiones, los LOS Neisseria síntomas del paciente y la extensión de la enfermedad.
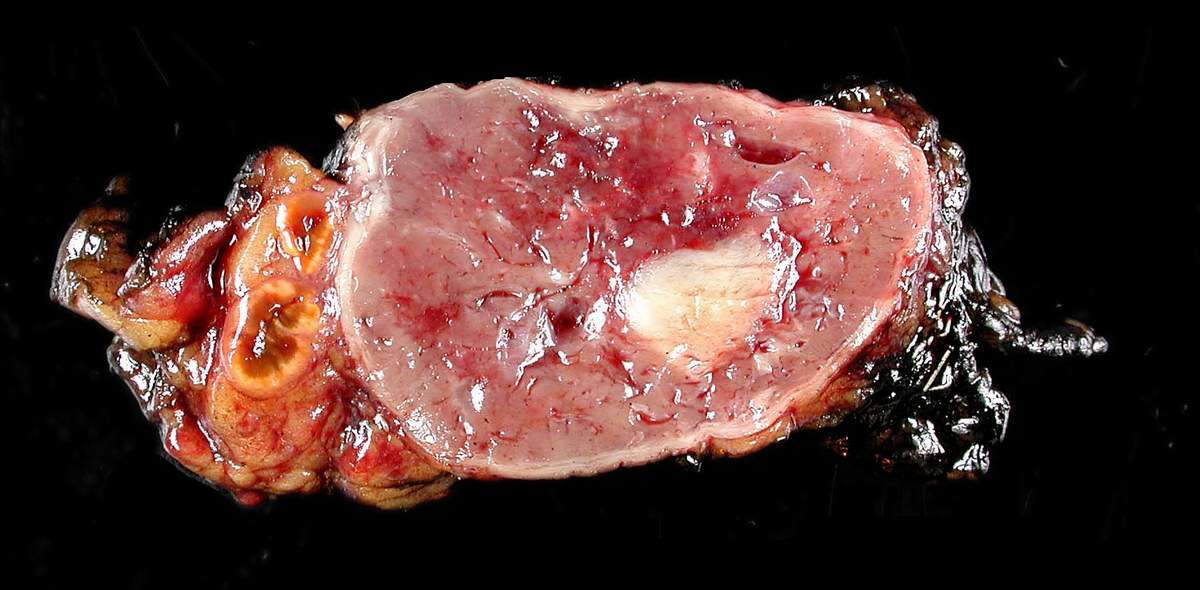
Feocromocitoma extirpado quirúrgicamente
Imagen: “Adrenal paraganglioma clinical Pheochromocytoma” por Michael Feldman. Licencia: CC BY 2.0