


A síndrome de Cowden, também conhecida como síndrome do hamartoma Hamartoma A focal malformation resembling a neoplasm, composed of an overgrowth of mature cells and tissues that normally occur in the affected area. Colorectal Cancer múltiplo, é uma doença hereditária autossómica dominante que se apresenta com crescimentos múltiplos e não cancerígenos em várias partes do corpo. A síndrome é classificada como uma síndrome de tumor Tumor Inflammation hamartoma Hamartoma A focal malformation resembling a neoplasm, composed of an overgrowth of mature cells and tissues that normally occur in the affected area. Colorectal Cancer homólogo de fosfatase e tensina (PTEN, pela sigla em inglês) causado por mutações no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics PTEN. Os doentes geralmente apresentam macrocefalia, triquilemomas (tumores do folículo piloso) e pápulas papilomatosas na boca. A síndrome de Cowden está associada a um aumento do risco de desenvolver cancro da mama, tiroide, cólon e do endométrio. O tratamento inclui vigilância de tumores associados e cirurgias profiláticas.
Last updated: Dec 15, 2025
A síndrome de Cowden é uma genodermatose autossómica dominante causada por uma mutação no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics PTEN e caracterizada por múltiplos hamartomas benignos em qualquer local do corpo, lesões mucocutâneas e macrocefalia.
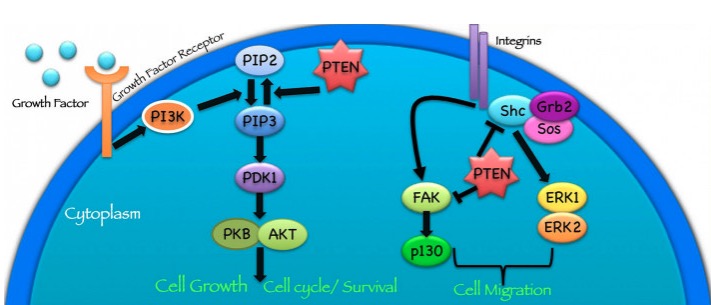
Função da proteína PTEN:
A proteína PTEN funciona de diferentes maneiras para suprimir a formação e disseminação de tumores.
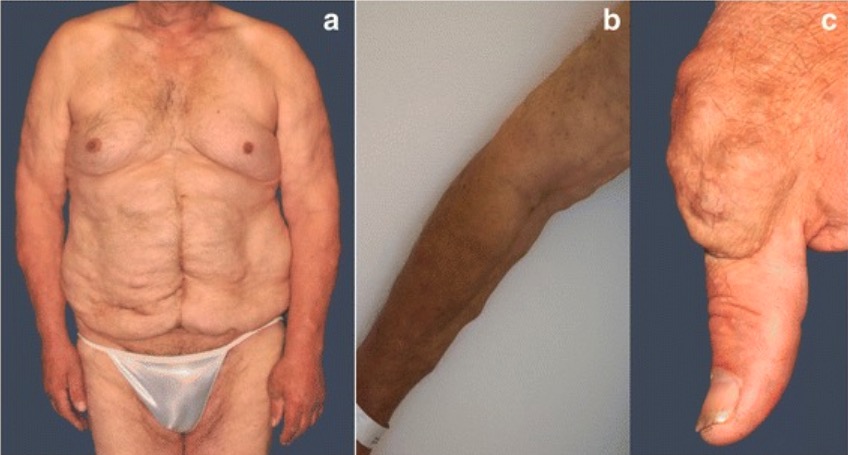
Vários tumores de tecidos moles no tronco (a), membros (b), e articulação basal do polegar esquerdo (c)
Imagem : “Fig. 2” por Steffen Pistorius et al. Licença: CC BY 4.0
Macrocefalia e glândula tiroideia difusamente aumentada em doente pediátrico com síndrome de Cowden
Imagem : “Diffusely enlarged thyroid gland” por Cláudia Patraquim et al. Licença: CC BY 2.0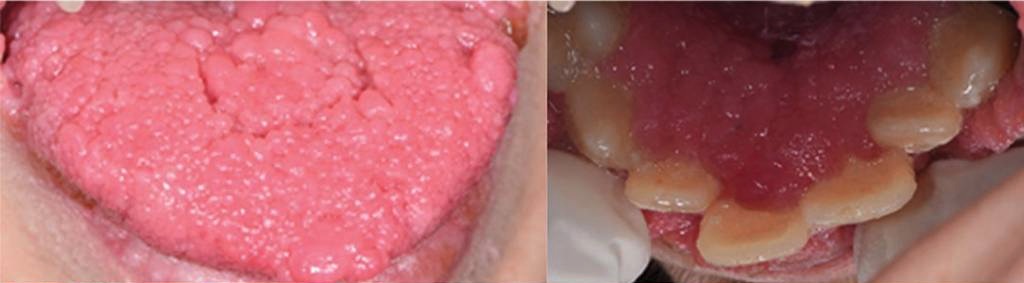
Esquerda: múltiplas lesões papilomatosas na língua
Direita: múltiplas lesões papilomatosas coalescentes cobrindo a região cervical dos dentes
Triquilemomas no osso malar direito de um doente com doença de hamartoma múltiplo
Imagem : “Trichilemmomas on the right malar bone” por Prashanthi Chippagiri et al. Licença: CC BY 3.0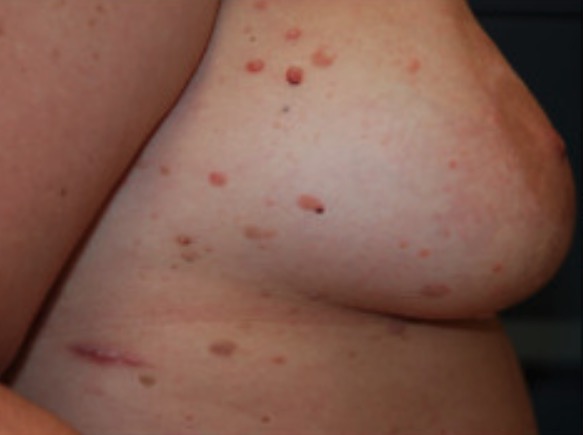
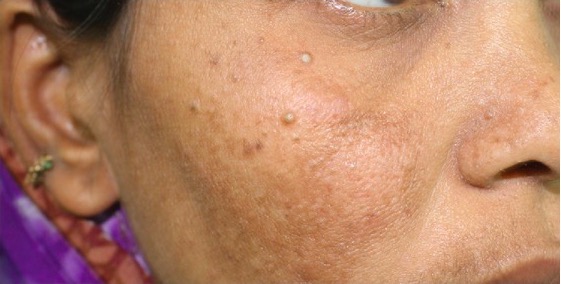
Queratoses seborreicas múltiplas num doente com carcinoma da mama bilateral de início precoce
Imagem: “Cutaneous findings of our case” por Laura Maria Pradella et al. Licença: CC BY 2.0, editado por Lecturio.Os exames laboratoriais são dirigidos pela apresentação clínica. A síndrome de Cowden pode apresentar-se com anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (por perdas GI), doença renal, doença da tiroide e lesões cutâneas).
Com base nas queixas à apresentação, as imagens para a síndrome de Cowden podem incluir:
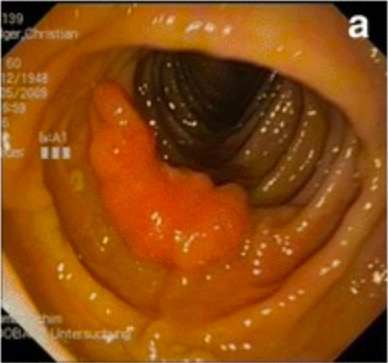
Hamartoma do cólon descendente em doente com síndrome de Cowden
Imagem: “Preoperative colonoscopic views” por Pistorius et al. Licença: CC BY 4.0, editado por Lecturio.