La policitemia vera es una neoplasia mieloproliferativa crónica caracterizada por la sobreproducción de eritrocitos. Además, también aumentan los LOS Neisseria recuentos de leucocitos y plaquetas, lo que diferencia la policitemia de la eritrocitosis que se observa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la hipoxia crónica y otras afecciones crónicas. Se presume que la policitemia vera tiene una base genética debido a mutaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen Janus kinase-2. La presentación clínica incluye síntomas relacionados con la enfermedad que pueden afectar varios sistemas de órganos. A veces, la enfermedad puede ser un hallazgo incidental durante las pruebas de laboratorio. El diagnóstico se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el análisis de sangre periférica y los LOS Neisseria hallazgos de la biopsia de médula ósea. El tratamiento es con flebotomía o terapia con medicamentos. El pronóstico generalmente es bueno y se prevé que la supervivencia del paciente mejore aún más con el amplio uso de nuevas terapias.
Last updated: Dec 15, 2025
La policitemia vera es una neoplasia mieloproliferativa crónica caracterizada por la sobreproducción de eritrocitosis, leucocitos y plaquetas. Esta tríada diferencia la policitemia vera de la eritrocitosis que se observa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la hipoxia crónica y otras afecciones.
La policitemia vera a menudo se diagnostica de manera incidental cuando un hemograma completo obtenido por otras razones revela un aumento de hemoglobina y hematocrito. Los LOS Neisseria pacientes también presentan síntomas o complicaciones relacionados con la enfermedad.
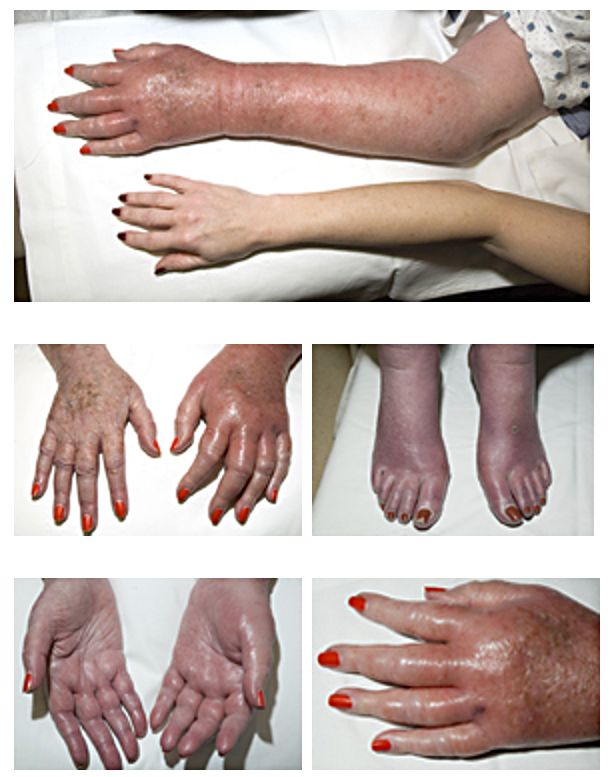
Eritromelalgia:
Sensación de dolor quemante en pies o manos acompañado de eritema, palidez o cianosis, en presencia de pulsos palpables
Se sospecha policitemia vera en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum pacientes con hallazgos físicos característicos y/o niveles elevados de hemoglobina y hematocrito en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum un hemograma completo.
Descartar causas secundarias de policitemia (niveles altos de eritropoyetina):
Otros hallazgos de laboratorio para policitemia vera primaria:
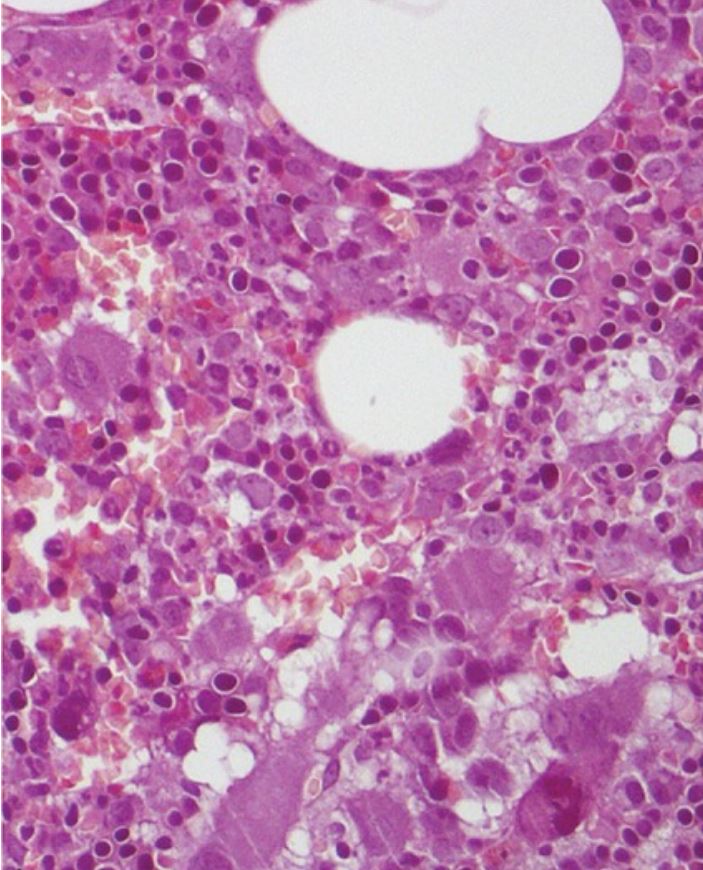
El análisis morfológico de la médula ósea no muestra cambios evidentes en la fibrosis de la médula ósea ni en el porcentaje de blastos, ni infiltrado linfoide significativo.
Imagen: “Erdheim-Chester Disease With Multiorgan Involvement, Following Polycythemia Vera: A Case Report” por Iurlo, A., et al. Licencia: CC BY 4.0, recortada por Lecturio.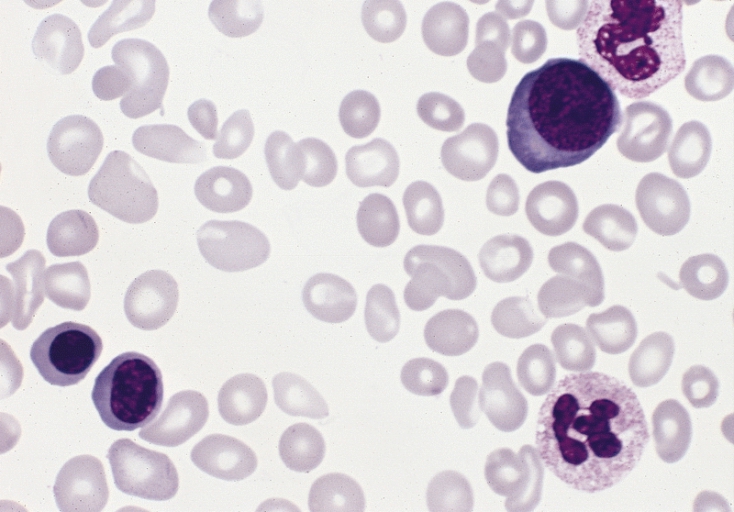
Frotis de sangre de diferente morfología de glóbulos rojos en un individuo con policitemia vera. Presencia de 3 precursores de hematíes y anisopoiquilocitosis de leve a moderada (tinción de Wright-Giemsa).
Imagen: “Polycythemia vera, blood smear” por The Armed Forces Institute of Pathology (AFIP). Licencia: Dominio Público