


A síndrome do X frágil (SXF), também conhecida como síndrome de Martin-Bell, é uma condição genética com hereditariedade ligada ao X. Podem ser afetados tanto meninos quanto meninas, mas a gravidade é muito maior nos meninos. As características típicas incluem um rosto comprido, testa e queixo proeminentes, orelhas grandes, pés chatos e testículos grandes após a puberdade nos meninos. A síndrome do X frágil é a causa mais MAIS Androgen Insensitivity Syndrome comum de deficiência intelectual hereditária e também está associada ao autismo. O teste genético confirma o diagnóstico. O tratamento visa melhorar os sintomas associados.
Last updated: Dec 15, 2025
A síndrome do X frágil (SXF) ocorre devido a uma mutação de perda de função no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics X frágil 1 do atraso mental (FMR1, pela sigla em inglês) no cromossoma X. O gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics FMR1 codifica a proteína X frágil do atraso mental (FMRP, pela sigla em inglês), que é importante no desenvolvimento do cérebro e das gonadas (testículos e ovários).
Mais MAIS Androgen Insensitivity Syndrome de 99% dos casos são devidos à expansão por repetição do trinucleotídeo instável (CGG, pela sigla em inglês) na região promotora do gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics FMR1:
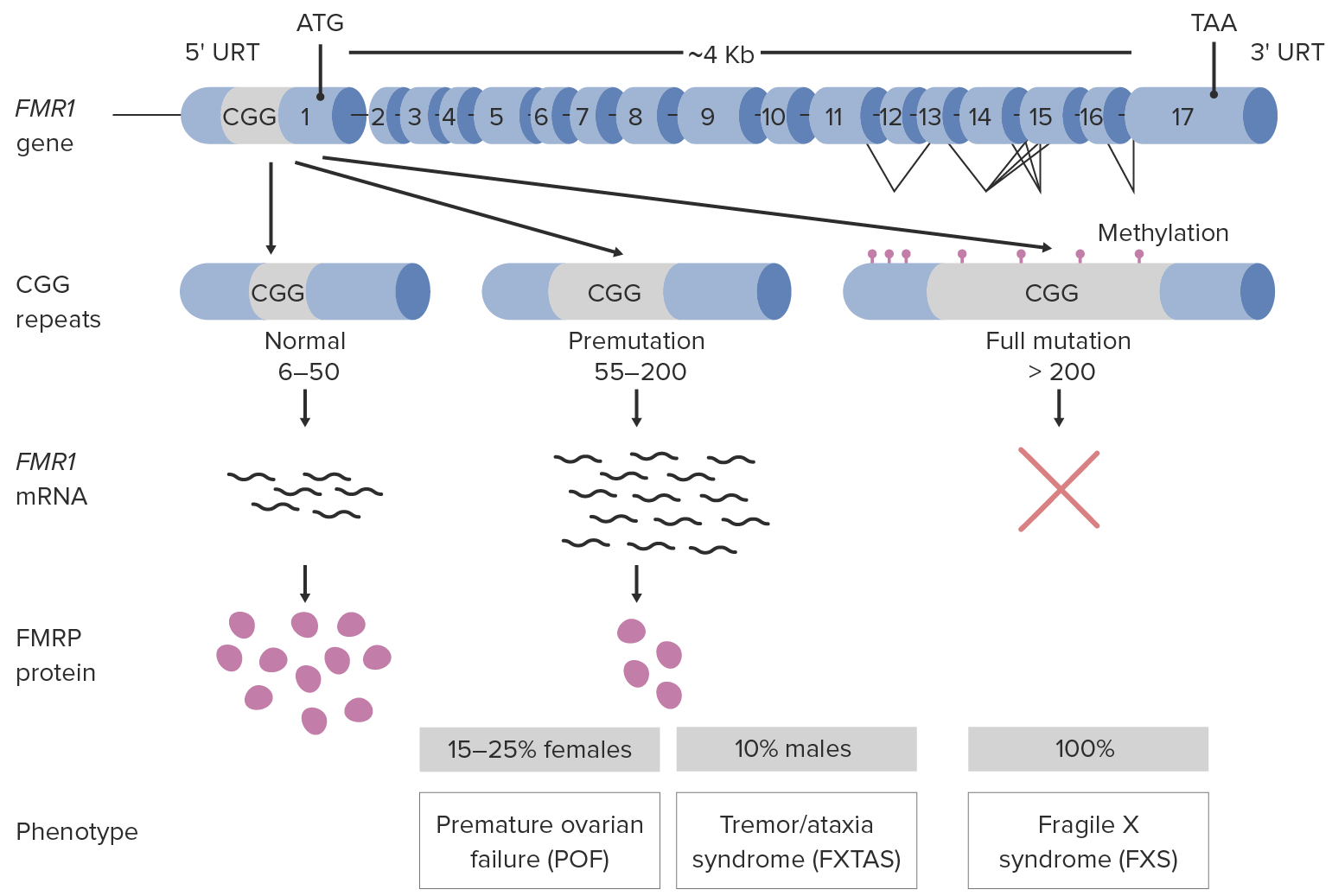
Fisiopatologia da síndrome do X frágil
FXTAS, pela sigla em inglês: Tremor/ataxia associada ao X frágil
O padrão de hereditariedade é dominante ligado ao X:
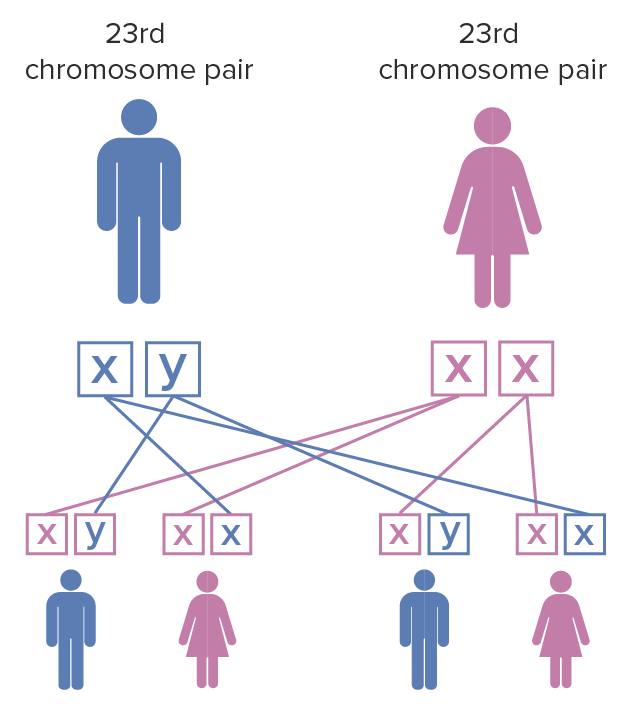
Padrão de hereditariedade da síndrome do X frágil:
A mãe tem um par de cromossomas X que sofreu mutação; o pai tem cromossomas X e Y não afetados. Quando o indivíduo do sexo feminino com a mutação do X frágil em ambos os cromossomas X tem filhos, cada filho tem 100% de probabilidade de herdar a doença. Os indivíduos do sexo feminino são menos severamente afetados pela síndrome do X frágil do que os do sexo masculino devido à inativação do cromossoma X.
As meninas e aqueles com pré-mutações têm manifestações clínicas variáveis e muitas vezes menos graves. Seguem-se as características clínicas de doentes com SXF clássico (mutação completa).
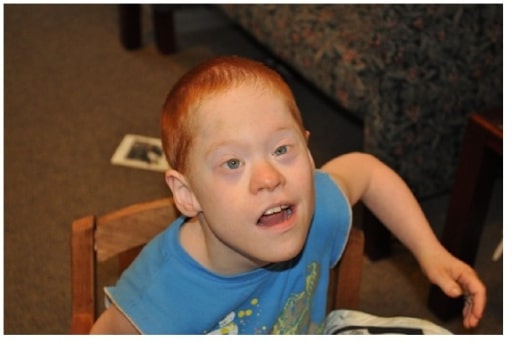
Menino com co-ocorrência de síndrome de Down e mutação completa SXF
Imagem: “Boy with cooccurrence DS and full mutation FXS” por Emory University, Department of Human Genetics, 2165 N. Decatur Road, Decatur, GA 30033, USA. Licença: CC BY 3.0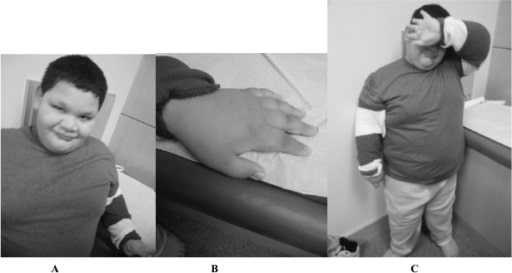
Um menino de 9 anos com mutação completa da SXF: Observar o rosto redondo e orelhas proeminentes (A), dedos curtos (B), e obesidade troncular (C).
Imagem: “Fragile X boy with the Prader-Willi phenotype” por Medical Investigation of Neurodevelopmental Disorders (M.I.N.D.) Institute, University of California Davis Health System, Sacramento, California, USA. Licença: CC BY 2.5Não há tratamento curativo para a SXF. O tratamento é adaptado aos sintomas presentes. A terapêutica de suporte inclui o seguinte:
As seguintes condições são diagnósticos diferenciais de SXF:
Listam-se as condições às quais se deve prestar atenção naqueles com pré-mutação da SXF.
