


O síndrome de Guillain-Barré (SGB), antes considerado um processo único de doença, é uma família de polineuropatias imunomediadas que ocorrem após infeções (por exemplo, com Campylobacter jejuni Campylobacter jejuni A species of bacteria that resemble small tightly coiled spirals. Its organisms are known to cause abortion in sheep and fever and enteritis in man and may be associated with enteric diseases of calves, lambs, and other animals. Campylobacter). O SGB típico é caracterizado por uma paralisia neuromuscular monofásica aguda, simétrica e com progressão ascendente. Se a paralisia atingir os músculos respiratórios, o SGB pode progredir para insuficiência respiratória, o que requer hospitalização prolongada. O tratamento é principalmente de suporte e pode exigir plasmaferese ou imunoglobulina IV.
Last updated: Dec 15, 2025
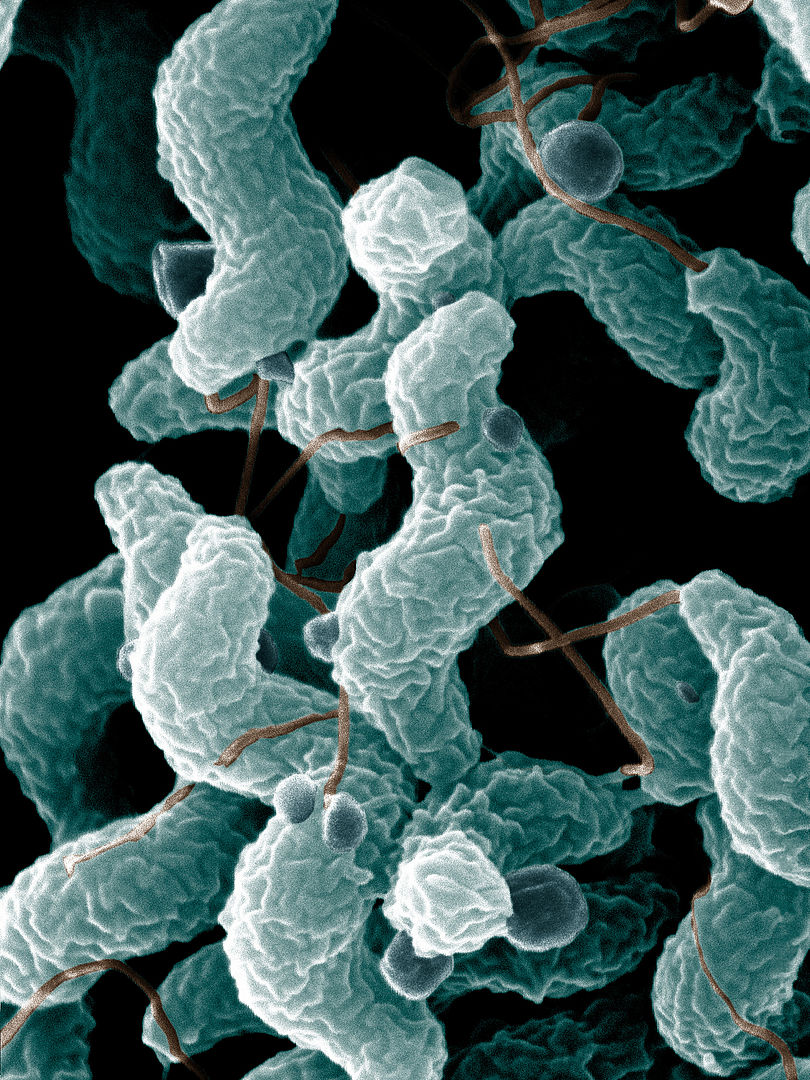
Campylobacter jejuni
Esta microscopia eletrónica mostra o organismo em forma de S com um único flagelo polar.
Os sintomas clínicos variam de acordo com o subtipo de SGB. Os doentes podem ter história de sintomas respiratórios ou gastrointestinais de 1 a 4 semanas antes do aparecimento de SGB.
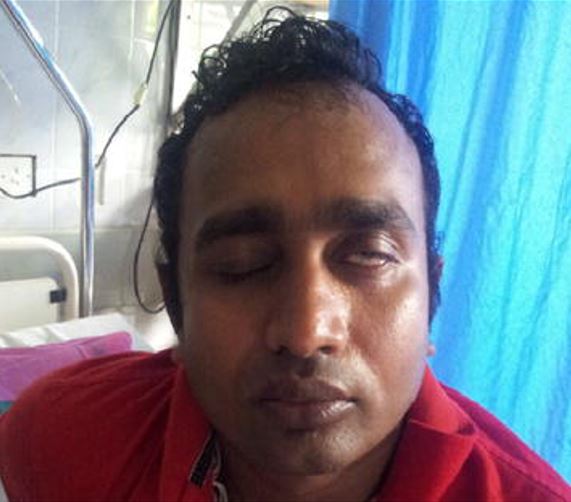
Sintomas clínicos da síndrome de Guillain-Barré:
Paralisia de Bell (paralisia do nervo facial) num indivíduo com SGB após uma infeção aguda por dengue
