


A síndrome de Alport, também denomidada nefrite hereditária, é um distúrbio genético causado por uma mutação nos genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure que codificam as cadeias alfa do colagénio tipo IV, resultando na alteração da produção de filamentos de colagénio tipo IV. Os doentes apresentam glomerulonefrite, hipertensão, edema Edema Edema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema, hematúria e proteinúria, bem como achados oculares (cataratas, retinopatia) e auditivos (perda auditiva neurossensorial). O diagnóstico é estabelecido com análise de urina, microscopia de urina e um painel de função renal. Uma biópsia renal a mostrar a delaminação característica da membrana basal glomerular pode ser usada para confirmar o diagnóstico. O tratamento da síndrome de Alport tem como objetivo limitar a progressão da doença com bloqueadores dos recetores da angiotensina e inibidores da enzima conversora da angiotensina. Os aparelhos auditivos são usados se perda auditiva associada à síndrome de Alport.
Last updated: Dec 15, 2025
A síndrome de Alport é um distúrbio genético decorrente de uma mutação nos genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure que codificam as cadeias alfa do colagénio tipo IV, localizadas principalmente no cristalino, na cóclea e nos rins (glomérulo).
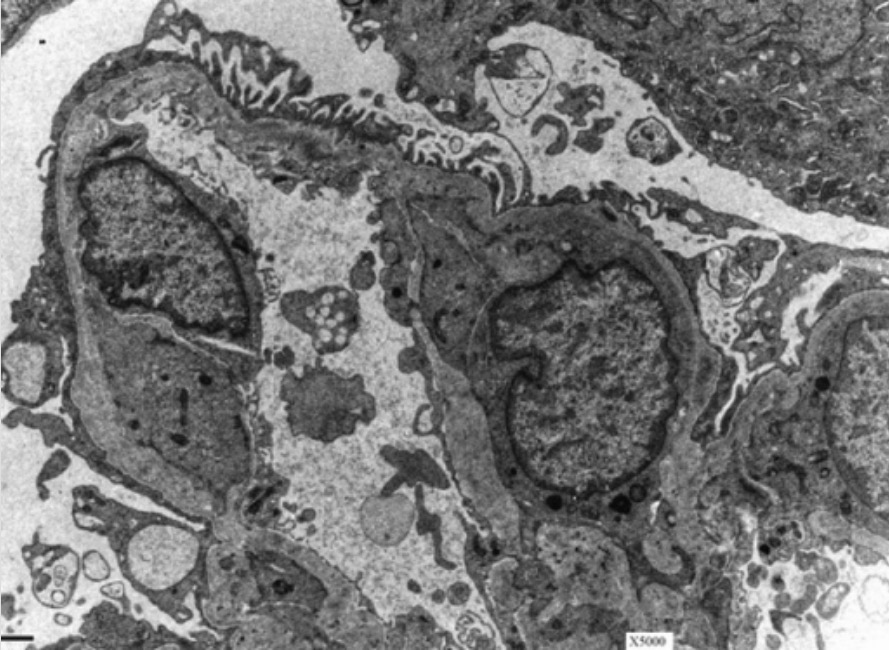
Membrana basal glomerular (MBG) em doente com síndrome de Alport:
A microscopia eletrónica de um rim mostra a aparência característica em cesta da MBG num doente com síndrome de Alport.
A síndrome de Alport geralmente apresenta-se com achados renais, oculares e auditivos.
Perda de audição neurosensorial:
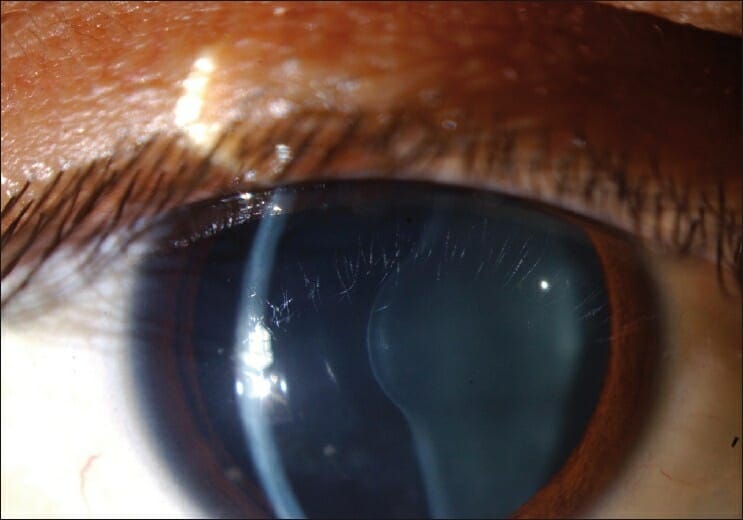
Lenticone anterior no olho de um indivíduo com síndrome de Alport
Imagem: “Figure 1 Anterior lenticonus in the right eye in a patient with Alport syndrome” por Ammar M Al-Mahmood, Samar A Al-Swailem, Abdulrahman Al-Khalaf, Ghada Y Al-Binali. Licença: CC BY 2.0É necessária uma história completa, incluindo história familiar e exame físico para fazer um diagnóstico.
O tratamento visa limitar a progressão da doença renal e proteinúria. Não está disponível, atualmente, nenhum tratamento definitivo.
