El síndrome de Alport, también llamado nefritis hereditaria, es un trastorno genético causado por una mutación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure que codifican las cadenas alfa del colágeno tipo IV, lo que resulta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la producción de hebras de colágeno tipo IV anormales. Los LOS Neisseria pacientes presentan glomerulonefritis, hipertensión, edema Edema Edema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema, hematuria Hematuria Presence of blood in the urine. Renal Cell Carcinoma y proteinuria Proteinuria The presence of proteins in the urine, an indicator of kidney diseases. Nephrotic Syndrome in Children, así como hallazgos oculares (cataratas, retinopatía) y auditivos (hipoacusia neurosensorial). El diagnóstico se establece con análisis de orina, microscopía de orina y un panel de función renal. Se puede utilizar una biopsia renal que muestre la división característica de la membrana basal glomerular para confirmar el diagnóstico. El tratamiento para el síndrome de Alport se centra en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum limitar la progresión de la enfermedad con bloqueadores de los LOS Neisseria receptores de angiotensina II e inhibidores de la enzima convertidora de angiotensina. Los LOS Neisseria audífonos se utilizan para la pérdida auditiva asociada con el síndrome de Alport.
Last updated: Dec 15, 2025
El síndrome de Alport es un trastorno genético que surge de una mutación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure que codifican las cadenas alfa del colágeno tipo IV, que se localiza principalmente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el cristalino, la cóclea y los LOS Neisseria riñones (glomérulo).
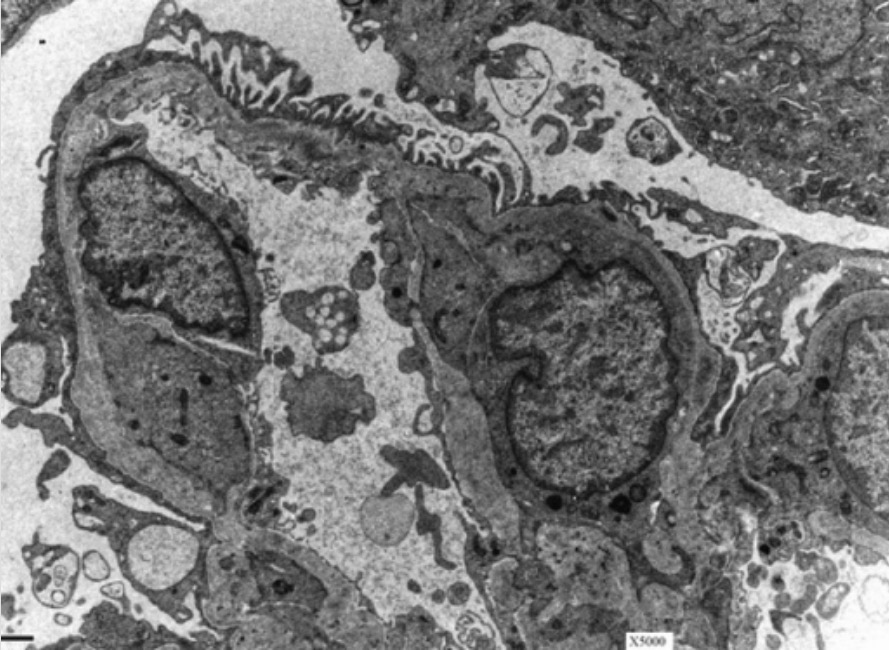
Membrana basal glomerular en un paciente con síndrome de Alport:
La microscopía electrónica de un riñón muestra el aspecto característico de tejido de cesta de la membrana basal glomerular en un paciente con síndrome de Alport.
El síndrome de Alport se presenta con mayor frecuencia con hallazgos renales, oculares y auditivos.
Hipoacusia neurosensorial:
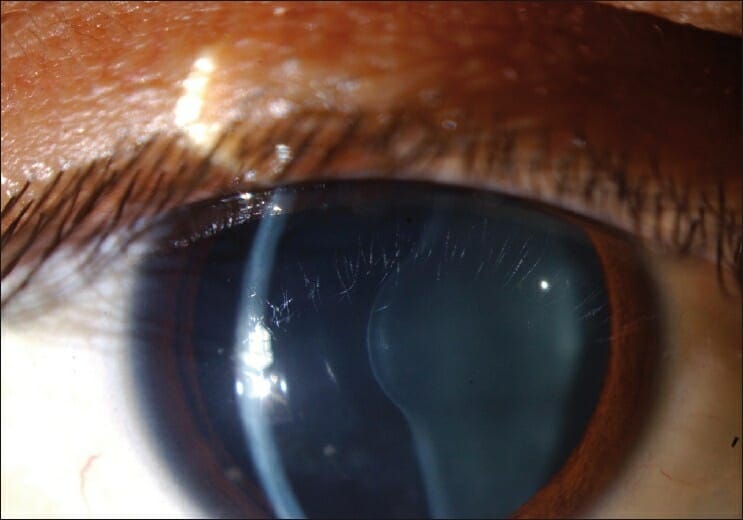
Lenticonus anterior en el ojo de un individuo con síndrome de Alport
Imagen: “Figure 1 Anterior lenticonus in the right eye in a patient with Alport syndrome” por Ammar M Al-Mahmood, Samar A Al-Swailem, Abdulrahman Al-Khalaf, Ghada Y Al-Binali. Licencia: CC BY 2.0Se requiere una toma de antecedentes completa, incluida los LOS Neisseria antecedentes familiares, y un examen físico para hacer un diagnóstico.
El tratamiento se centra en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum limitar la progresión de la enfermedad renal y la proteinuria Proteinuria The presence of proteins in the urine, an indicator of kidney diseases. Nephrotic Syndrome in Children. Actualmente no se dispone de un tratamiento definitivo.