


A Poliarterite nodosa (PAN) é uma vasculite necrosante, sistémica, principalmente de vasos médios. A maioria dos casos de PAN são idiopáticos. Os fatores etiológicos da PAN secundária são a infeção por hepatite B e C e a leucemia de células pilosas. A PAN apresenta sintomas e sinais sistémicos tais como fadiga, perda de peso, febre, fraqueza, artralgia, hipertensão e edema dos membros. Uma vez que se trata de uma doença multissistémica que afeta a pele, os nervos, os músculos, os rins e os vasos sanguíneos que abastecem o coração e o trato gastrointestinal, a apresentação clínica depende dos órgãos envolvidos. O diagnóstico é feito com base na história, exame físico e resultados de testes laboratoriais e confirmado por biópsia ou angiografia. O tratamento depende da gravidade e extensão da doença. O método de tratamento primário é com glucocorticoides orais; agentes imunossupressores também podem ser necessários para os doentes com doença moderada a grave.
Last updated: Dec 15, 2025
A poliarterite nodosa (PAN) é uma vasculite sistémica, principalmente associada aos vasos médios, caracterizada por lesões inflamatórias necrotizantes. Esta condição acaba por resultar em microaneurismas que levam a hemorragias, trombose, isquemia e enfartes de órgãos.
Os doentes com PAN apresentam tipicamente um envolvimento multissistémico, com sinais e sintomas sistémicos. No entanto, existe também uma forma limitada à pele denominada PAN cutânea.
| Sistema | Apresentação clínica |
|---|---|
| Renal |
|
| Cutâneo |
|
| Sistema nervoso |
|
| Muscular |
|
| Gastrointestinal |
|
| Cardiovascular |
|
| Reprodutivo |
|
| Oftalmológico |
|
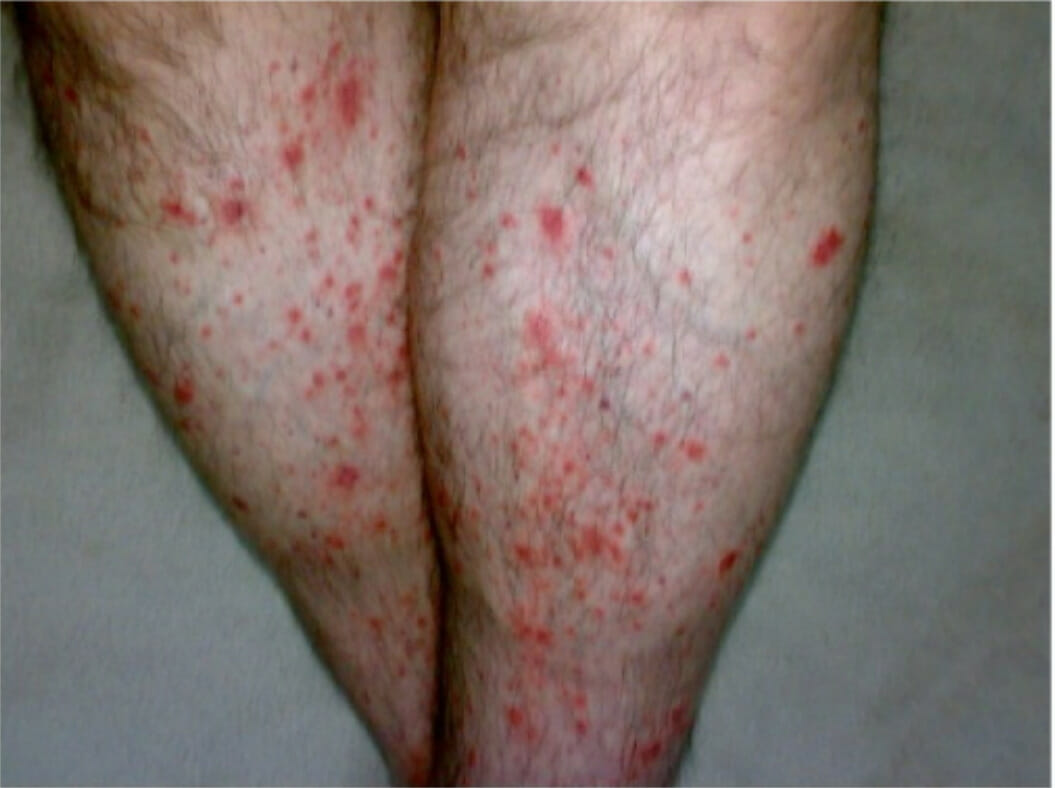
Púrpura palpável bilateral como manifestação cutânea de PAN
Imagem: “Bilateral palpable purpura in lower extremities” por gan Y, Doğru A, Şencan H, Şahin M, Ercan Tunç Ş.. Licença: CC BY 4.0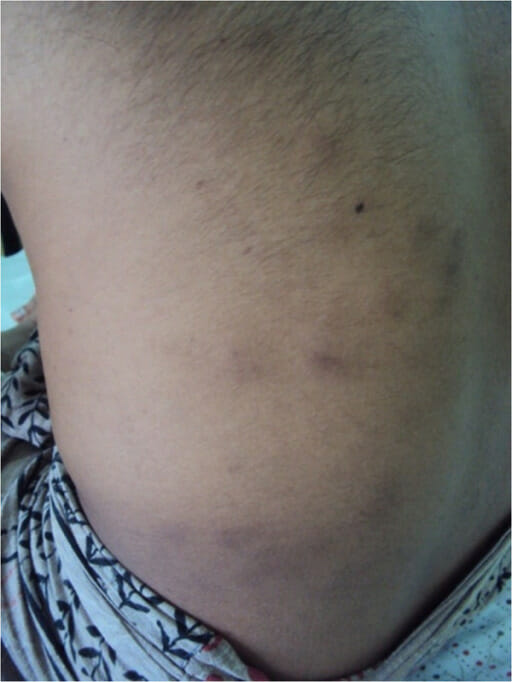
PAN associado à infeção pelo VHC.
Está presente uma erupção cutânea do tipo paniculite com nódulos duros e dolorosos
O diagnóstico de PAN é feito com base na história, exame físico e resultados de testes Testes Gonadal Hormones laboratoriais e deve ser confirmado por biópsia, sempre que possível.
Os resultados laboratoriais são geralmente inespecíficos e podem ser utilizados estudos para avaliar o envolvimento de sistemas de órgãos e excluir outros diagnósticos prováveis.
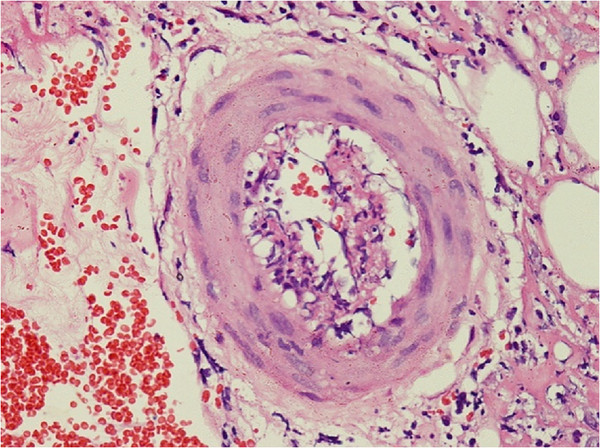
A biopsia cutânea mostra infiltração linfocítica e neutrofílica perivascular com necrose fibrinoide da parede do vaso e extravasamento leucocitoclástico e de glóbulos vermelhos num indivíduo com poliarterite nodosa.
Imagem: “Skin biopsy shows perivascular lymphocytic and neutrophilic infiltration” por Rodrigo D et al. Licença: CC BY 2.0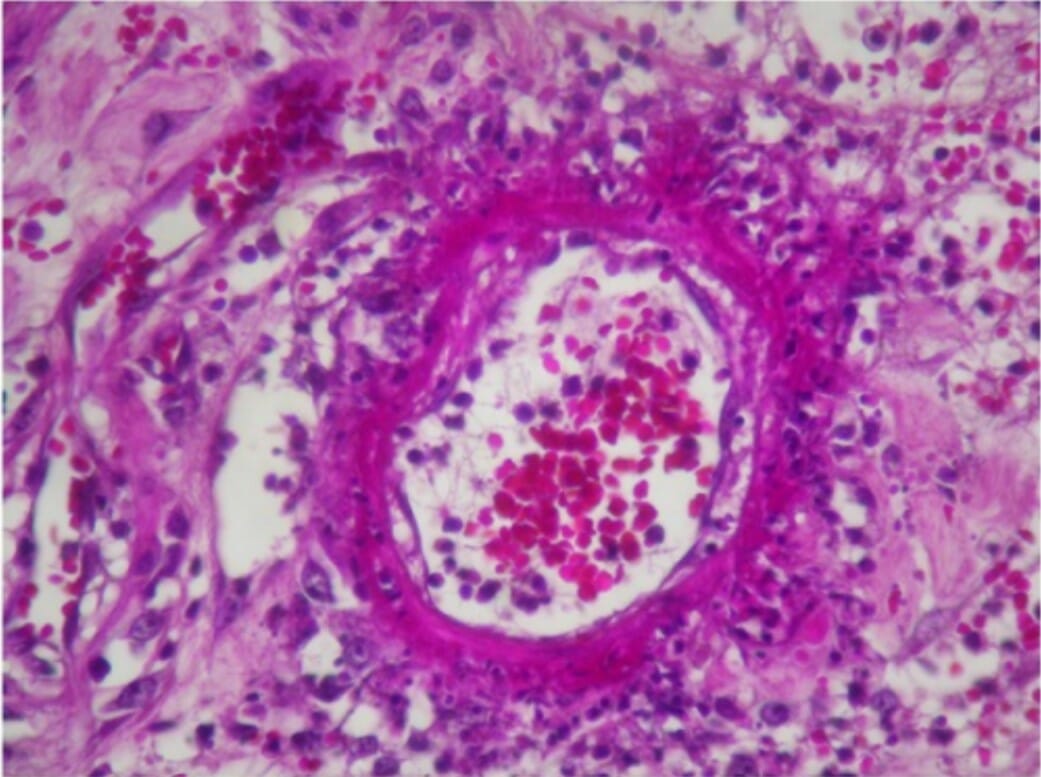
Colite isquémica devido à vasculite PAN:
exame histológico do cólon
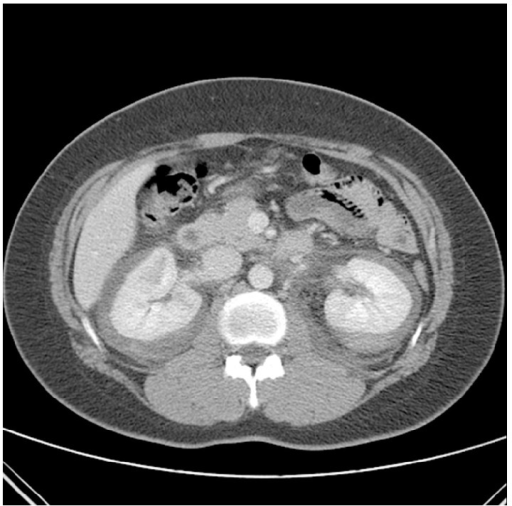
TC contrastada com hemorragia perirrenal bilateral espontânea (complicação rara) como a apresentação inicial de PAN, num indivíduo com dor nos flancos e hipovolemia
Imagem: “Contrast enhanced CT on the day of admission. CT revealed nonspecific spontaneous bilateral perirenal hemorrhage and delayed enhancing lesion of renal medulla” por Choi HI, Kim YG, Kim SY, Jeong da W, Kim KP, Jeong KH, Lee SH, Moon JY. Licença: CC BY 3.0A avaliação da gravidade da doença é útil para determinar a melhor opção de tratamento.
O tratamento depende da gravidade e extensão da doença e deve ser orientado por um especialista (reumatologista). O método de tratamento primário é com glucocorticoides orais. Os agentes imunossupressores também podem ser necessários em doentes com doença moderada a grave.
PAN ligeira:
PAN de moderada a severa:
Monitorização da PAN:
O prognóstico depende da gravidade da doença e da resposta ao tratamento.
