


A miocardiopatia arritmogénica ventricular direita (MAVD) é uma doença hereditária do músculo cardíaco que afeta o ventrículo direito (VD); pode causar perturbações do ritmo e morte súbita cardíaca (MSC). A doença resulta de mutações nos genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure que codificam as proteínas desmossómicas envolvidas na adesão de célula a célula. Os pacientes sintomáticos desenvolvem palpitações, arritmias que levam a síncope, dispneia ou toracalgia. O diagnóstico baseia-se na apresentação clínica, no ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG), no ecocardiograma e nos exames de imagem. O tratamento visa prevenir a MSC e as arritmias sintomáticas. O tratamento inclui a colocação de cardioversor-desfibrilhador implantável (CDI), ablação por radiofrequência para corrigir arritmias e farmacoterapia antiarrítmica. A prevenção da progressão da doença com mudanças no estilo de vida é importante e o transplante cardíaco pode ser necessário após 15 anos.
Last updated: Jan 21, 2026
A miocardiopatia arritmogénica ventricular direita (MAVD) é uma doença genética do músculo cardíaco caracterizada pela substituição fibroadiposa do tecido miocárdico ventricular direito.
A apresentação clínica da miocardiopatia arritmogénica ventricular direita é variável e pode permanecer silenciosa durante décadas, tornando difícil o seu reconhecimento.
Os detalhes do Relatório de 2017 do American College of Cardiology/ American Heart Association American Heart Association A voluntary organization concerned with the prevention and treatment of heart and vascular diseases. Heart Failure Task Force e da Heart Rhythm Society (ACC/AHA/ HRS HRS Hepatorenal syndrome (HRS) is a potentially reversible cause of acute kidney injury that develops secondary to liver disease. The main cause of hrs is hypovolemia, often as a result of forced diuresis or drainage of ascites. This leads to renal vasoconstriction resulting in hypoperfusion of the kidneys. Hepatorenal Syndrome) estão além deste âmbito. O relatório define disfunção global ou regional e alterações estruturais com medidas ecocardiográficas detalhadas da via de saída do VD.
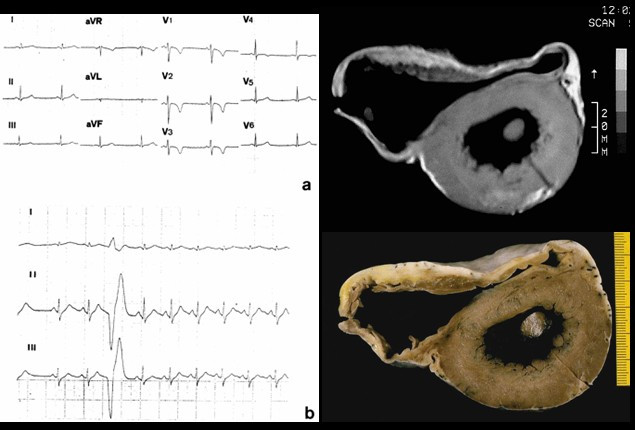
Esquerda: ECG de 12 derivações a) Ondas T invertidas em V1-V4 e b) Batimentos ectópicos ventriculares.
direita: secção transversal do coração com dilatação do ventrículo direito, aneurismas anterior e posterior
Os principais objetivos no tratamento da miocardiopatia arritmogénica ventricular direita são prevenir arritmias que ameacem a vida e atrasar a progressão da doença.
