La miocardiopatía arritmogénica del ventrículo derecho es un trastorno hereditario del músculo cardíaco que afecta al AL Amyloidosis ventrículo derecho; puede causar alteraciones del ritmo y muerte súbita cardíaca. Este trastorno es el resultado de mutaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure que codifican las proteínas desmosómicas que intervienen en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la adhesión de célula a célula. Los LOS Neisseria pacientes sintomáticos desarrollan palpitaciones, arritmias que provocan síncopes, disnea o dolor Dolor Inflammation torácico. El diagnóstico se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la presentación clínica, ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG), ecocardiográficos y de estudios de imagenología. El objetivo del tratamiento es prevenir la muerte súbita cardíaca y las arritmias sintomáticas. El tratamiento incluye la colocación de un desfibrilador cardioversor implantable, ablación por radiofrecuencia para corregir las arritmias y medicamentos antiarrítmicos. La prevención de la progresión de la enfermedad con cambios en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el estilo de vida es importante y el trasplante cardíaco puede ser necesario en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum caso de insuficiencia cardíaca terminal.
Last updated: Jan 21, 2026
La miocardiopatía arritmogénica del ventrículo derecho es una enfermedad genética del músculo cardíaco que se caracteriza por la sustitución fibrograsa del miocardio del ventrículo derecho.
La presentación clínica de la miocardiopatía arritmogénica del ventrículo derecho es variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables y puede no presentarse síntomas durante décadas, lo que dificulta su reconocimiento.
Los LOS Neisseria detalles del Informe del 2017 del Grupo de Trabajo del American College of Cardiology/ American Heart Association American Heart Association A voluntary organization concerned with the prevention and treatment of heart and vascular diseases. Heart Failure y the Heart Rhythm Society (ACC/AHA/ HRS HRS Hepatorenal syndrome (HRS) is a potentially reversible cause of acute kidney injury that develops secondary to liver disease. The main cause of hrs is hypovolemia, often as a result of forced diuresis or drainage of ascites. This leads to renal vasoconstriction resulting in hypoperfusion of the kidneys. Hepatorenal Syndrome) están fuera del alcance de este artículo. El informe define la disfunción global o regional y las alteraciones estructurales basándose en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum mediciones ecocardiográficas detalladas del tracto de salida del ventrículo derecho.
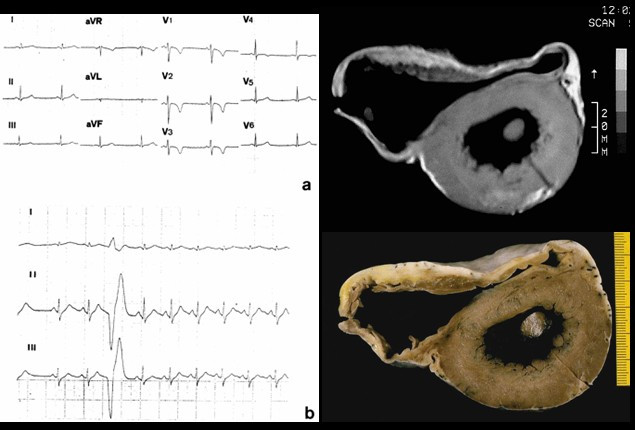
Izquierda: el electrocardiograma de 12 derivaciones muestra (a) ondas T invertidas en V1-V4 y (b) latidos ectópicos ventriculares.
Derecha: sección transversal del corazón con la dilatación del ventrículo derecho, aneurismas anteriores y posteriores
Los LOS Neisseria objetivos principales en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el tratamiento de la miocardiopatía arritmogénica del ventrículo derecho es prevenir las arritmias potencialmente mortales y frenar la progresión de la enfermedad.