


As malformações de Chiari (CMs, pela sigla em inglês) são um grupo de condições do sistema nervoso central (CNS, pela sigla e inglês) caracterizadas pelo subdesenvolvimento da fossa craniana posterior com subsequente protrusão de estruturas neurais através do buraco magno. Existem 4 tipos de CM, sendo o tipo I o mais MAIS Androgen Insensitivity Syndrome comum. As cefaleias são o sintoma mais MAIS Androgen Insensitivity Syndrome comum. O diagnóstico é feito por achados clínicos e confirmado por ressonância magnética (RM). O tratamento é cirúrgico, baseado na descompressão da fossa posterior e restauração do fluxo do CNS. O prognóstico depende do tipo de malformação.
Last updated: Dec 15, 2025
As malformações de Chiari (CMs) são um grupo de doenças definidas por défices estruturais no cérebro e na medula espinhal que limitam o espaço da fossa posterior, o que força as estruturas cerebelares a projetarem-se através do buraco magno.
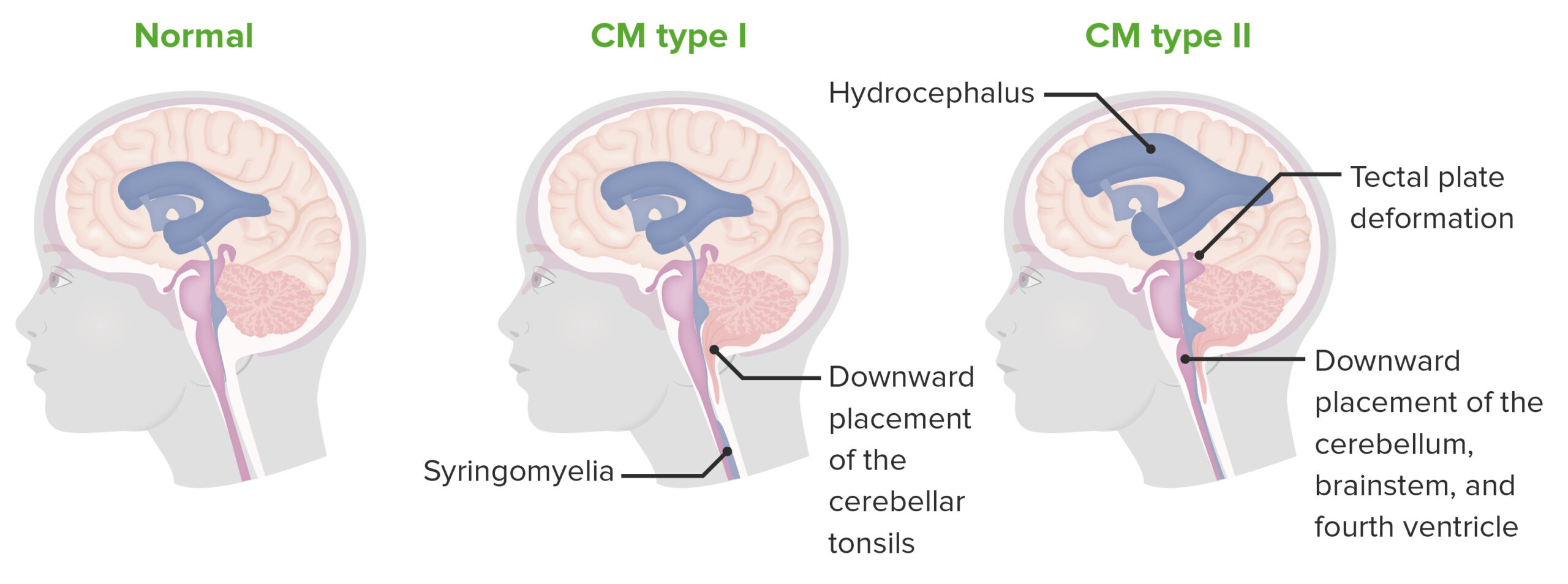
Chiari tipo I e II
As malformações de Chiari são caracterizadas por uma fossa craniana posterior subdesenvolvida, com subsequente protrusão de estruturas neurais através do buraco magno. O tipo 1 é caracterizado por herniação apenas das amígdalas cerebelares, enquanto o tipo 2 envolve mais estruturas.
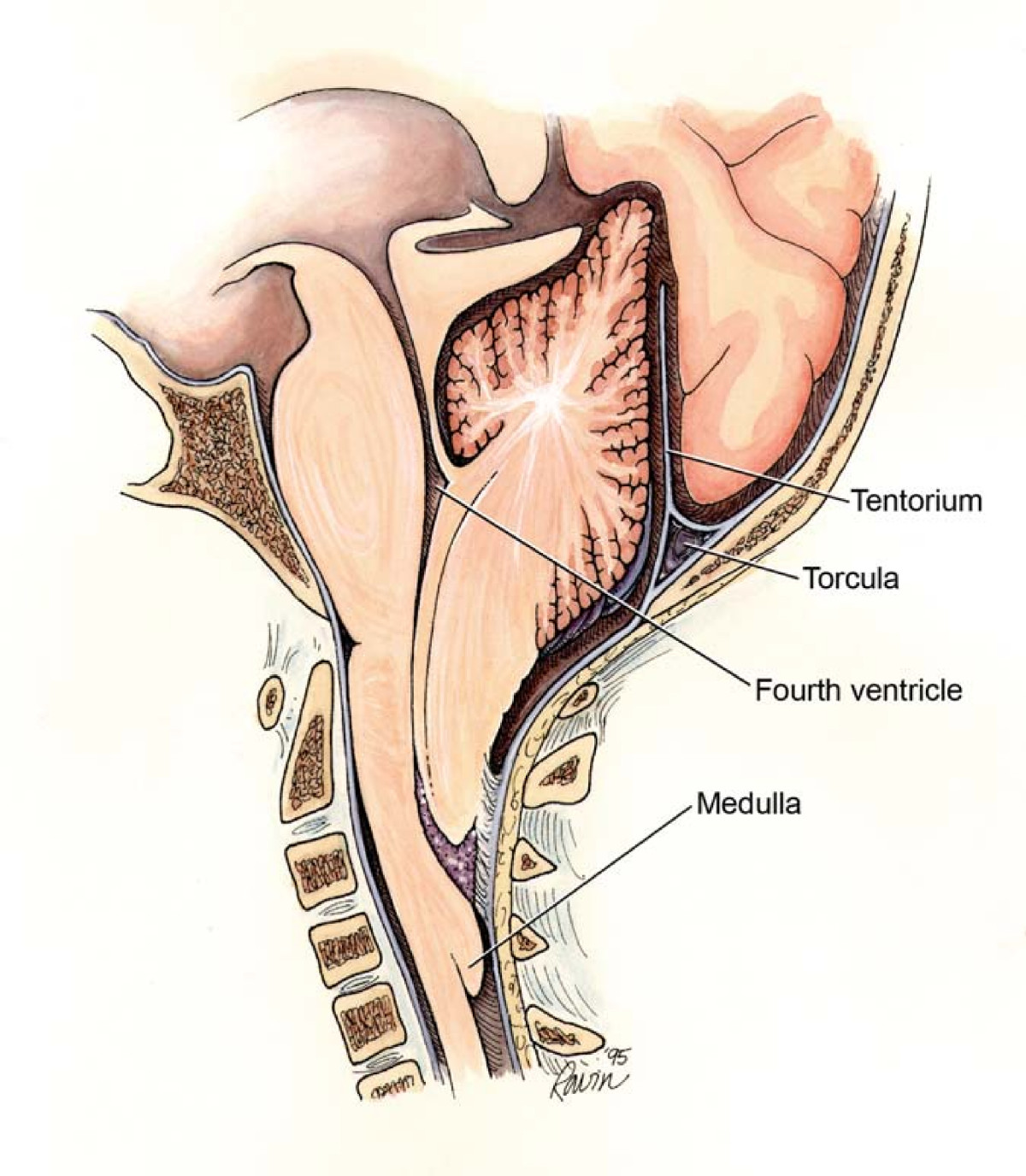
Malformação de Chiari II que mostra os pontos de potencial obstrução que levam aos diferentes subtipos de hidrocefalia
Imagem : “Chiari2” por Rekate HL. Licença: CC BY 2.0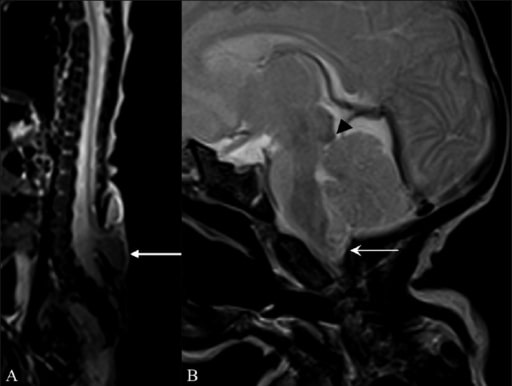
Achados de ressonância magnética (RM) sugestivos de malformação de Arnold-Chiari II
RM da coluna inteira sagital T2W (A) mostra um meningomielocelo (seta) oposto às vértebras L5 e S1. Ressonância magnética sagital T2W do cérebro (B) mostra uma pequena fossa craniana posterior, com herniação do vermis cerebelar e das amígdalas (seta) através do buraco magno com bico tectal (ponta de seta).
Incidência:
Condições associadas:
Os sintomas neurológicos são causados por:
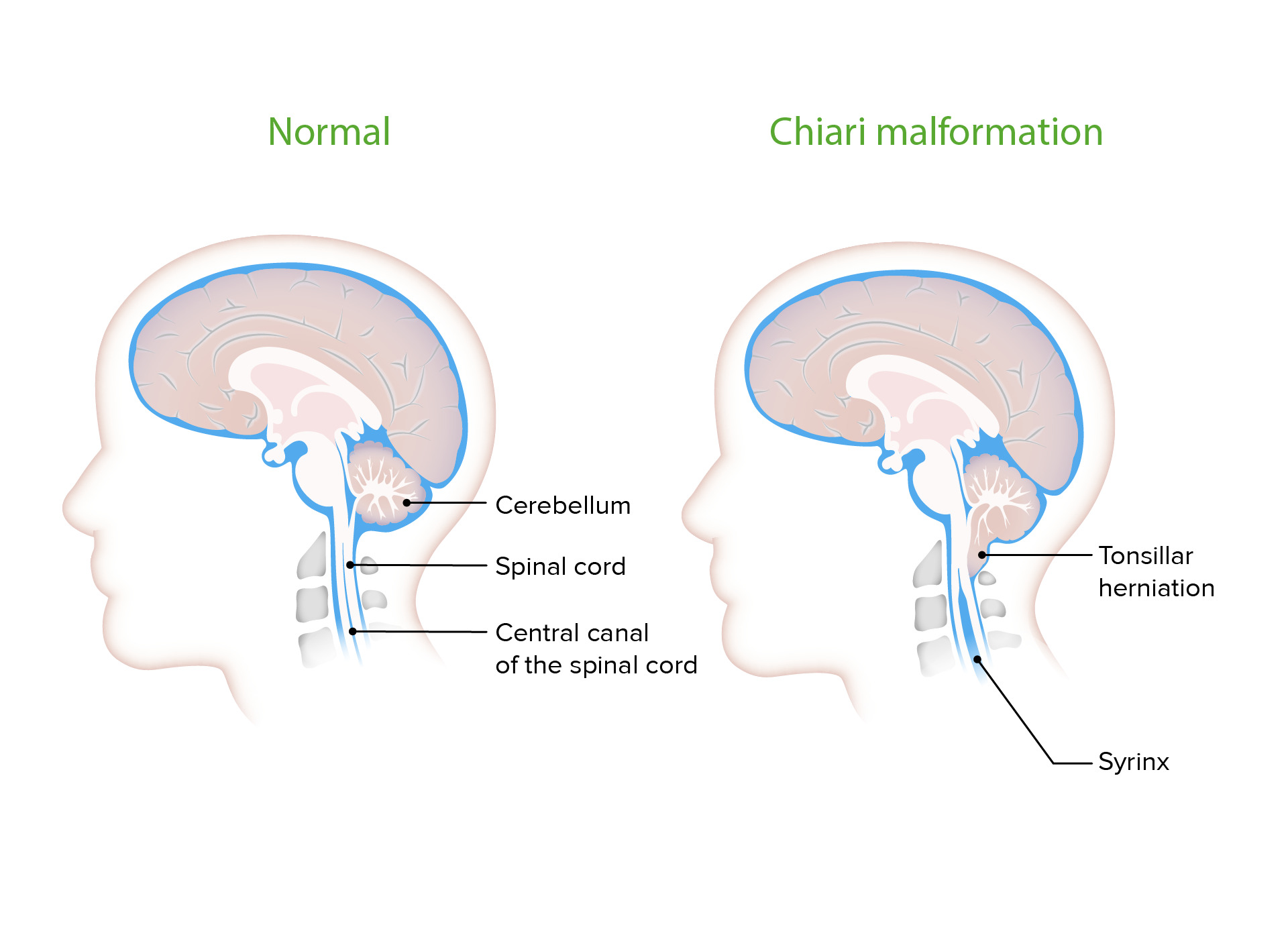
Siringe com malformação de Chiari
Cavidades preenchidas com líquido cefalorraquidiano (CSF, pela sigla em inglês) são frequentemente observadas com CMs. Causam sintomatologia frequentemente associada à malformação, pressionando o tecido neural circundante.
Sintomas:
Sinais físicos:
Sintomas:
Sinais físicos:
Há alta mortalidade infantil com este tipo.
Sintomas:
Sinais físicos:
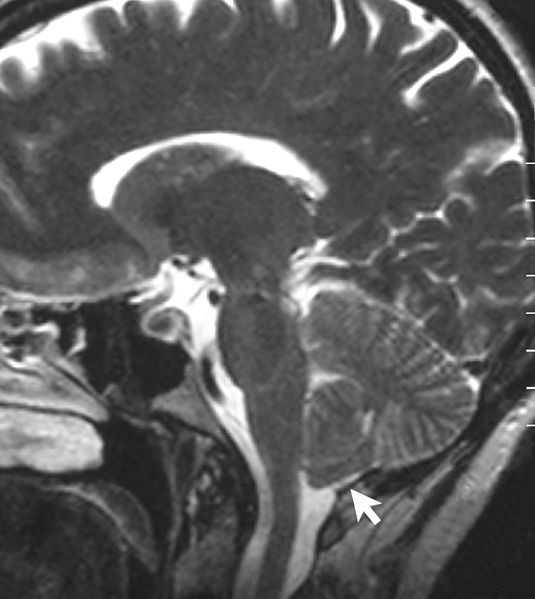
Ressonância magnética que mostra um CM tipo I. Observar a hérnia amidgalina.
Imagem : “Sagittal MRI scan of brain of patient with Chiari malformation” por Raymond F Sekula Jr et al. Licença: CC BY 2.0