A histiocitose das células de Langerhans (HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have "hair-like projections" visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia) é uma doença neoplásica rara e idiopática das células dendríticas causada por mutações somáticas dos genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and StructureBRAF, MAP2K1, RASRASRenal artery stenosis (RAS) is the narrowing of one or both renal arteries, usually caused by atherosclerotic disease or by fibromuscular dysplasia. If the stenosis is severe enough, the stenosis causes decreased renal blood flow, which activates the renin-angiotensin-aldosterone system (RAAS) and leads to renovascular hypertension (RVH).Renal Artery Stenosis e ARAF. Os sintomas generalizados podem incluir febre, fadiga e perda de peso. A HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have "hair-like projections" visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar apresenta dispneia, dor pleurítica torácica e uma tosse não produtiva. As manifestações da HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have "hair-like projections" visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia não pulmonar dependem do órgão envolvido (e.g., dor óssea, endocrinopatias). A abordagem diagnóstica envolve a colheita de uma história completa e a realização do exame físico, com testesTestesGonadal Hormones laboratoriais de base e imagiologia (e.g., ecografia). A tomografia por emissão de positrões (PET-TC) de corpo inteiro deteta a atividade da doença e a biópsia das lesões confirma o diagnóstico. O tratamento está dependente da extensão da doença. As opções terapêuticas incluem o tratamento local (e.g., curetagem da lesão se doença óssea, radioterapia) e a terapêutica sistémica (e.g., agentes quimioterápicos, terapêutica alvo).
A histiocitose de células de Langerhans (HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia) é uma doença neoplásica idiopática rara das células dendríticas (células de Langerhans), que estão envolvidas na apresentação do antigénio às células T.
A incidência é maior nas populações leucodérmicas.
O tabagismo intenso aumenta o risco de HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar isolada.
Classificação
A Histiocyte Society e a OMS têm ambas classificações para as doenças histiocíticas.
A classificação da Histiocyte Society inclui tanto as doenças malignas como as benignas.
A classificação da OMS aborda apenas as doenças malignas.
Classificação da Histiocyte Society
A Histiocyte Society divide as perturbações histiocíticas em 5 categorias, com base no seguinte:
Características clínicas
Características histológicas
Características imunofenotípicas
Características moleculares
Tabela: Classificação da Histiocyte Society
Grupo da doença histiocítica
Perturbações
Grupo Langerhans (L)
Histiocitose de células de Langerhans (HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia)
Doença de Erdheim-Chester (DEC)
Misto de HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia/DEC
Histiocitose de células indeterminadas
Xantogranuloma juvenil extracutâneo
Grupo cutâneo e mucocutâneo (C)
Xantogranuloma juvenil
Xantogranuloma adulto
Doença cutânea de Rosai-Dorfman
Outras doenças histiocíticas localizadas na pele ou superfícies mucosas que não satisfazem os critérios de diagnóstico da HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia
Grupo da doença de Rosai-Dorfman (R)
Doença de Rosai-Dorfman
Histiocitoses não-cutâneas diversas que não satisfazem os critérios de diagnóstico da LCH
Grupo de histiocitose maligna (M)
Histiocitoses malignas primárias com envolvimento de múltiplos sistemas
Histiocitoses malignas secundárias a outros linfomas e leucemias
Grupo de linfohistiocitose hemofagocítica (H)
Linfohistiocitose hemofagocítica primária (LHF)
Síndromes de ativação macrofágica (SAMSAMAnterior displacement of the mitral valve during systole.Hypertrophic Cardiomyopathy)
Etiologia e Fisiopatologia
Etiologia
A HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia é causada por mutações somáticas de genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure que regulam a via de sinalização MAPK/ERK, como por exemplo:
BRAF
MAP2K1
RASRASRenal artery stenosis (RAS) is the narrowing of one or both renal arteries, usually caused by atherosclerotic disease or by fibromuscular dysplasia. If the stenosis is severe enough, the stenosis causes decreased renal blood flow, which activates the renin-angiotensin-aldosterone system (RAAS) and leads to renovascular hypertension (RVH).Renal Artery Stenosis
ARAF
Fisiopatologia
Mutações genéticas → ativação da via MAPK → expansão clonal dos precursores mieloides
As células dendríticas proliferam e acumulam-se devido à estimulação imunitária contínua.
As células dendríticas com proliferação anormal, depois infiltram-se em ≥ 1 órgão → manifestações clínicas
A HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pode apresentar-se como uma doença de sistema único ou multissistema.
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia de sistema único:
Os sintomas sistémicos estão frequentemente ausentes.
Multisistema HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia:
Envolve ≥ 2 órgãos
A combinação de órgãos envolvidos varia, mas pode incluir:
Osso
Pele
Hipotalâmico-hipófise
Gânglios linfáticos
Pulmões
SNC
Fígado
Baço
Mucosa oral
Em crianças < 3 anos, a HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia é frequentemente uma doença multissistémica.
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar
Associada ao tabagismo (90% dos doentes são ex-fumadores ou fumadores atuais)
Os sintomas constitucionais (e.g., febre, perda de peso) são observados em 20% dos casos.
LCH não-pulmonar
Os sinais e sintomas dependem do número e da localização dos locais envolvidos.
Tabela: Apresentação clínica da HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia por localização
Sistema
Manifestações (podem incluir qualquer uma das seguintes):
Geral
Diminuição do apetite
Mal-estar
Palidez cutânea
Infeções frequentes
Febre
Hematomas fáceis
Perda de peso
Atraso no crescimento (crianças)
Pulmonar
Dor pleurítica torácica
Tosse não produtiva
Dispneia
Hemoptise
Cianose
Osso
EdemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema ou tumefação, indolor ou dolorosa
Fraturas frequentes
Dor óssea
Exoftalmia (cavidade orbital)
Secreção auricular, perda de audição (osso temporal)
Cutâneo
Pode ocorrer uma vasta gama de erupções:
EczemaEczemaAtopic dermatitis, also known as eczema, is a chronic, relapsing, pruritic, inflammatory skin disease that occurs more frequently in children, although adults can also be affected. The condition is often associated with elevated serum levels of IgE and a personal or family history of atopy. Skin dryness, erythema, oozing, crusting, and lichenification are present. Atopic Dermatitis (Eczema)
Pápulas castanhas-púrpura
Lesões ulcerativas
Pode também ser pustular, purpúrica, petequial ou vesicular
Pode ser escamosa, com crosta, ou com secreções
Descoloração e endurecimento das unhas
Qualquer parte do corpo pode ser afetada.
Oral
Hipertrofia gengival
Doença periodontal
Massa intra-oral
Ulceração das mucosas
Dentes soltos
Hematologia
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Leucopenia
Trombocitopenia
Trato gastrointestinal
Dor abdominal
Vómito
Diarreia
Má absorção
Sangue nas fezes
Fígado
Distensão abdominal
Icterícia
Prurido
Hepatomegalia e/ou esplenomegalia
Endócrino
Polidipsia
Poliúria
Atraso do crescimento e da puberdade
Aumento de peso (envolvimento da glândula hipofisária)
Sintomas de hipotiroidismo (envolvimento da glândula tiroideia)
Sistema nervoso central
Movimentos corporais descoordenados
AtaxiaAtaxiaImpairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions.Ataxia-telangiectasia
Disartria
Perturbações visuais
Nistagmo
Dor de cabeça
Vertigens
Convulsões
Alterações comportamentais
Alterações da memória e défice de aprendizagem
Défices hormonais hipofisários (e.g., diabetesDiabetesDiabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance.Diabetes Mellitus insípida central)
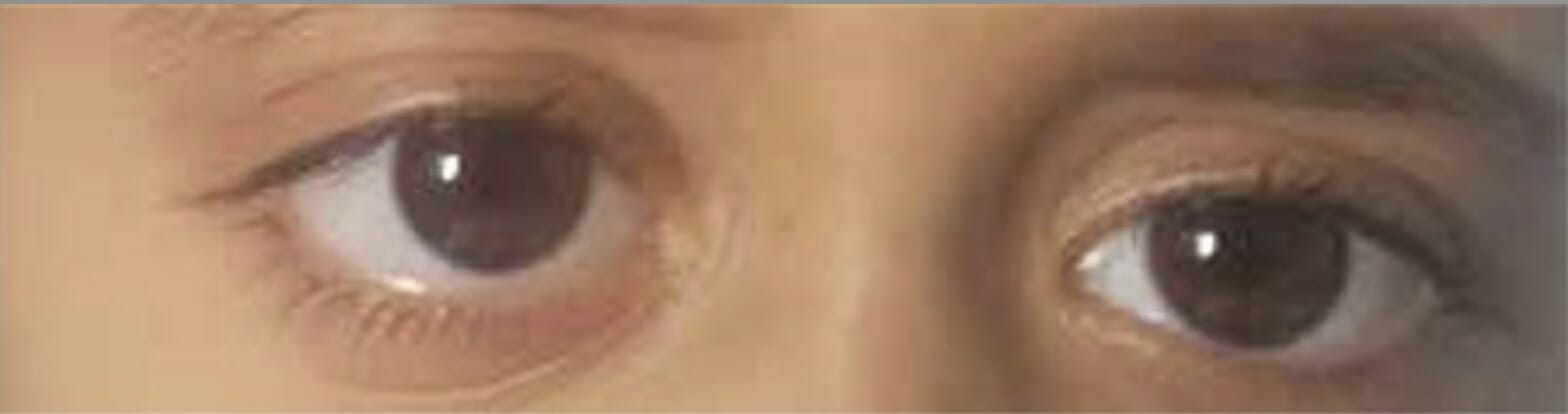
Exoftalmia do lado direito como resultado do envolvimento da parede orbital pela histiocitose de células de Langerhans
Imagem: “Proptosis due to Langerhans Cell” por Turkish Journal of Ophthalmology. Licença: CC BY 2.5
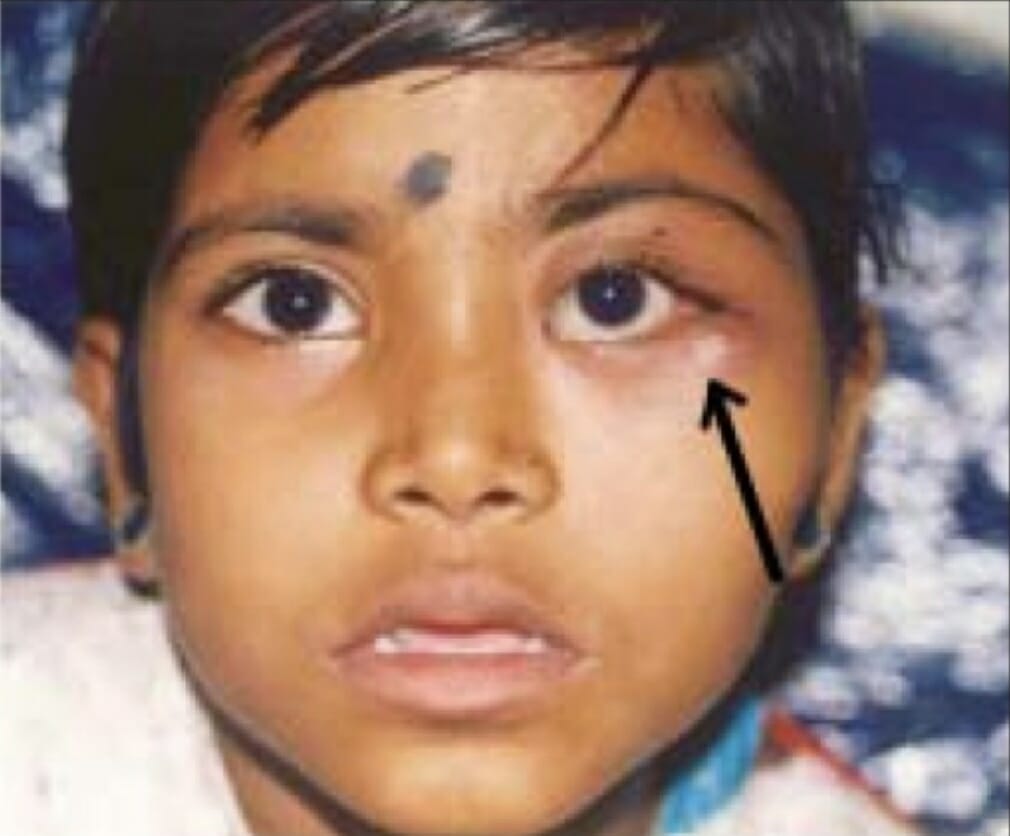
Massa mal definida (seta) na parede orbital lateral devido à histiocitose de células de Langerhans
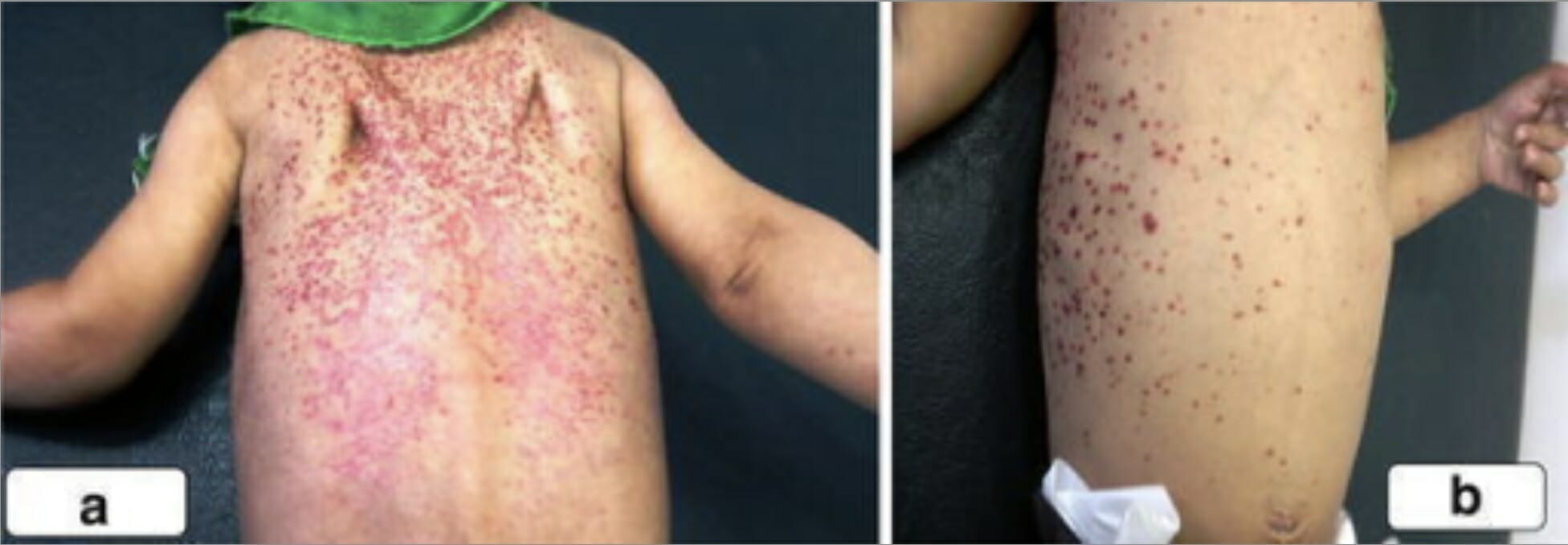
Imagem: “LCH presented with ill-defined mass in lateral orbital wall” por Department of Ophthalmology, Sri Sankaradeva Nethralaya, Guwahati. Licença: CC BY 2.0Erupção escamosa, eritematosa nas costas, ombros e tórax ito de um rapaz com histiocitose de células de LangerhansImagem: “A scaly, erythematous rash on the back of the second boy spread to his shoulders (a) and upper chest wall (b)” por Unit of Pediatric Surgery, Al Diwaniya General Teaching Hospital, Al Qadisiya, Iraq. Licença: CC BY 4.0
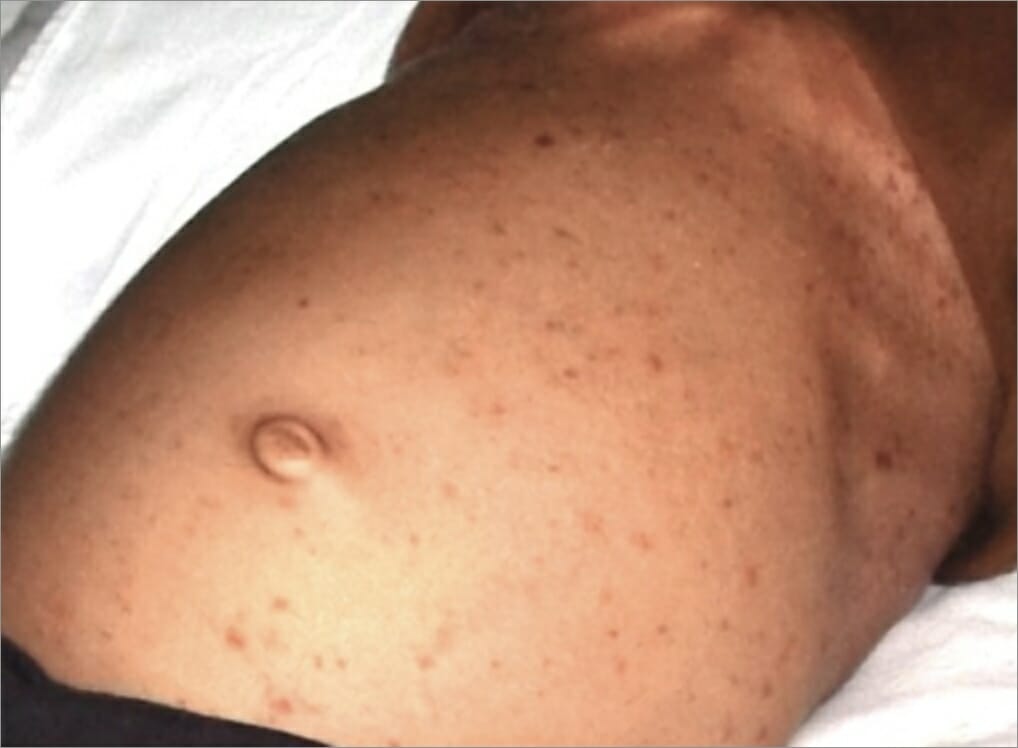
Erupção papular no estômago de um indivíduo como manifestação de histiocitose de células de Langerhans
Imagem: “After hospitalization” por Department of Oral Medicine and Radiology, Jaipur Dental College, Jaipur, Rajasthan, India. Licença: CC BY 3.0
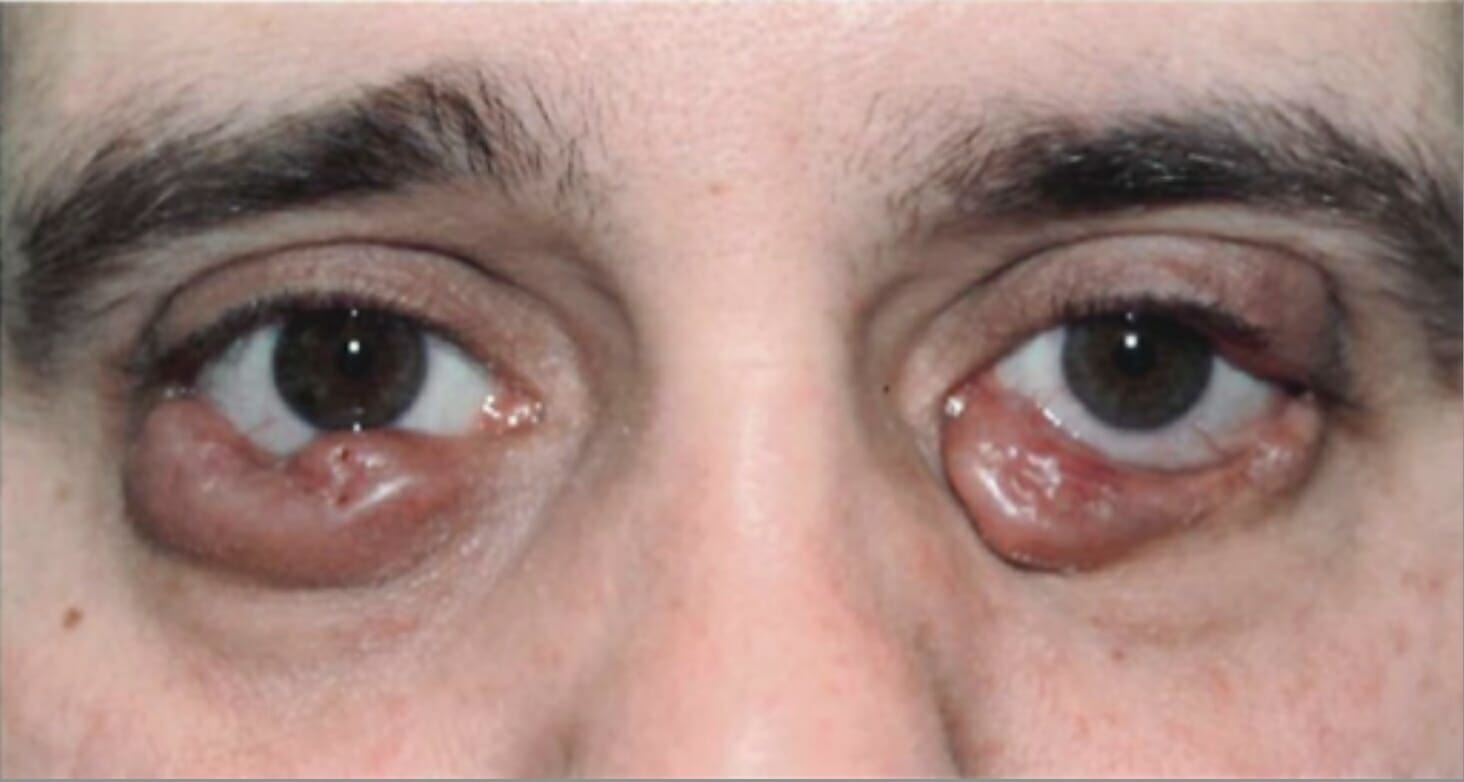
Crescimento bilateral das pálpebras inferiores devido à histiocitose de células de Langerhans
Imagem: “Before radiation treatment” por King’s College London. Licença: CC BY 3.0
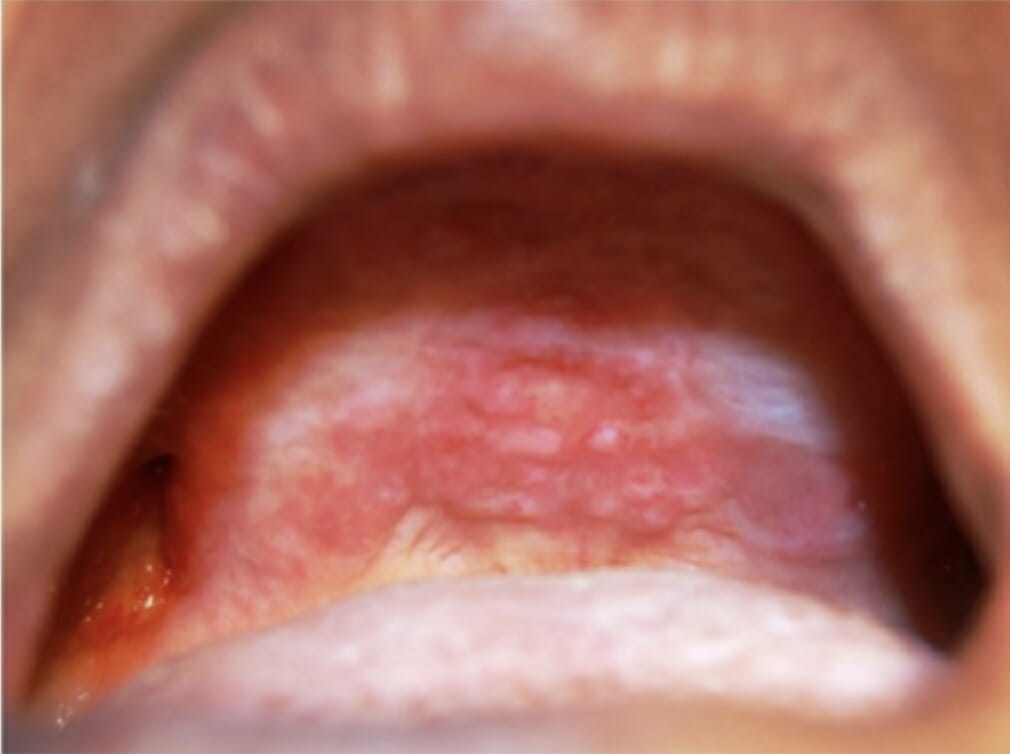
Lesões orais no palato duro e na crista alveolar maxilar como manifestação da histiocitose de células de Langerhans
Imagem: “Oral lesions in the hard palate and maxillary alveolar ridge” por Department of Oral Medicine and Radiology, M S Ramaiah Dental College and Hospital, Bangaluru. Licença: CC BY 2.5
Complicações
As complicações dependem dos sistemas de órgãos envolvidos, mas podem incluir:
Pneumotórax
Doença vascular pulmonar
Disfunção pulmonar crónica
Fratura óssea patológica
Cirrose hepática
Perfuração intestinal
Hemorragia GI
Doenças malignas secundárias, como a leucemia linfoblástica aguda
Cegueira (raro)
Diagnóstico
Avaliação laboratorial
Os estudos laboratoriais demonstram geralmente resultados consistentes com os sistemas de órgãos envolvidos e ajudam a excluir outros diagnósticos (a lista não é exaustiva):
Análises gerais:
Hemograma completo (com diferencial) → avaliar o envolvimento da medula óssea:
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Trombocitopenia
Leucopenia
Tempo de protrombina (TP) e tempo de tromboplastina parcial ativada (TTPa) → coagulopatia (particularmente se envolvimento hepático)
Eletrólitos → pode demonstrar perturbações relacionadas com doenças endócrinas
Painel hepatobiliar → ↑ se envolvimento hepatobiliar
A avaliação endócrina detalhada pode incluir:
Hormona estimulante da tiroide (TSH, pela sigla em inglês), T4T4The major hormone derived from the thyroid gland. Thyroxine is synthesized via the iodination of tyrosines (monoiodotyrosine) and the coupling of iodotyrosines (diiodotyrosine) in the thyroglobulin. Thyroxine is released from thyroglobulin by proteolysis and secreted into the blood. Thyroxine is peripherally deiodinated to form triiodothyronine which exerts a broad spectrum of stimulatory effects on cell metabolism.Thyroid Hormones livre
Prolactina e fator de crescimento semelhante à insulina 1 (IGF-1)
CortisolCortisolGlucocorticoids sérico, hormona adrenocorticotrópica (ACTH, pela sigla em inglês)
Osmolalidade do soro e da urina
Hormona folículo-estimulante (FSHFSHA major gonadotropin secreted by the adenohypophysis. Follicle-stimulating hormone stimulates gametogenesis and the supporting cells such as the ovarian granulosa cells, the testicular sertoli cells, and leydig cells. Fsh consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle, pela sigla em inglês) e hormona luteinizante (LHLHA major gonadotropin secreted by the adenohypophysis. Luteinizing hormone regulates steroid production by the interstitial cells of the testis and the ovary. The preovulatory luteinizing hormone surge in females induces ovulation, and subsequent luteinization of the follicle. Luteinizing hormone consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle, pela sigla em inglês)
Testosterona (homens)
Estrogénio (mulheres)
Imagiologia e estudos auxiliares adicionais
Radiografia de base na apresentação inicial:
Radiografia torácica → imagem inicial se sintomas pulmonares:
Nódulos mal definidos ou espiculados
Padrão reticulonodular bilateral difuso (zonas médias e superiores)
Quistos da zona superior ou favos de mel
Volume pulmonar conservado
Poupa dos ângulos costofrénicos
Radiografia do osso/articulação afetada: as lesões são tipicamente osteolíticas (aspeto “perfurado”).
PET-TC de corpo inteiro com fluorodesoxiglicose (FDG):
A HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia é ávida por FDG, pelo que a PET-TC deteta o envolvimento de órgãos.
Superior à radiografia, TC ou RMN na deteção de lesões ósseas HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia
Realizada em doentes com ≥ 2 anos
Recomendada para estadiamento
Outros estudos dependem dos locais de envolvimento:
RMN cerebral com contraste de gadolínio:
Se envolvimento do SNC
Deteta massas intra-cranianas
Útil na avaliação da glândula hipofisária
TC:
Pode avaliar o trato GI, fígado, baço, mediastino e envolvimento pulmonar
Na HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar, observam-se opacidades e quistos reticulonodulares (zonas médias e superiores).
Pode mostrar redução da capacidade difusão se anomalias obstrutivas, restritivas ou mistas
Ecografia: pode ser usada na avaliação do fígado/tiroide
Achados da PET na histiocitose de células pulmonares nodulares de Langerhans: As imagens da TC torácica nos paineis superior e inferior esquerdos mostram múltiplos nódulos pulmonares. As imagens PET correspondentes nos paineis superiores e inferiores à direita mostram captação por PET. Os nódulos pulmonares maiores (setas nas imagens TC) demonstram uma captação intensa por PET, enquanto os outros nódulos (setas mais pequenas nas imagens TC) são PET-negativos.
Imagem: “PET findings in nodular PLCH” por Division of Pulmonary and Critical Care Medicine, Mayo Clinic, Rochester, MN, USA. Licença: CC BY 2.0
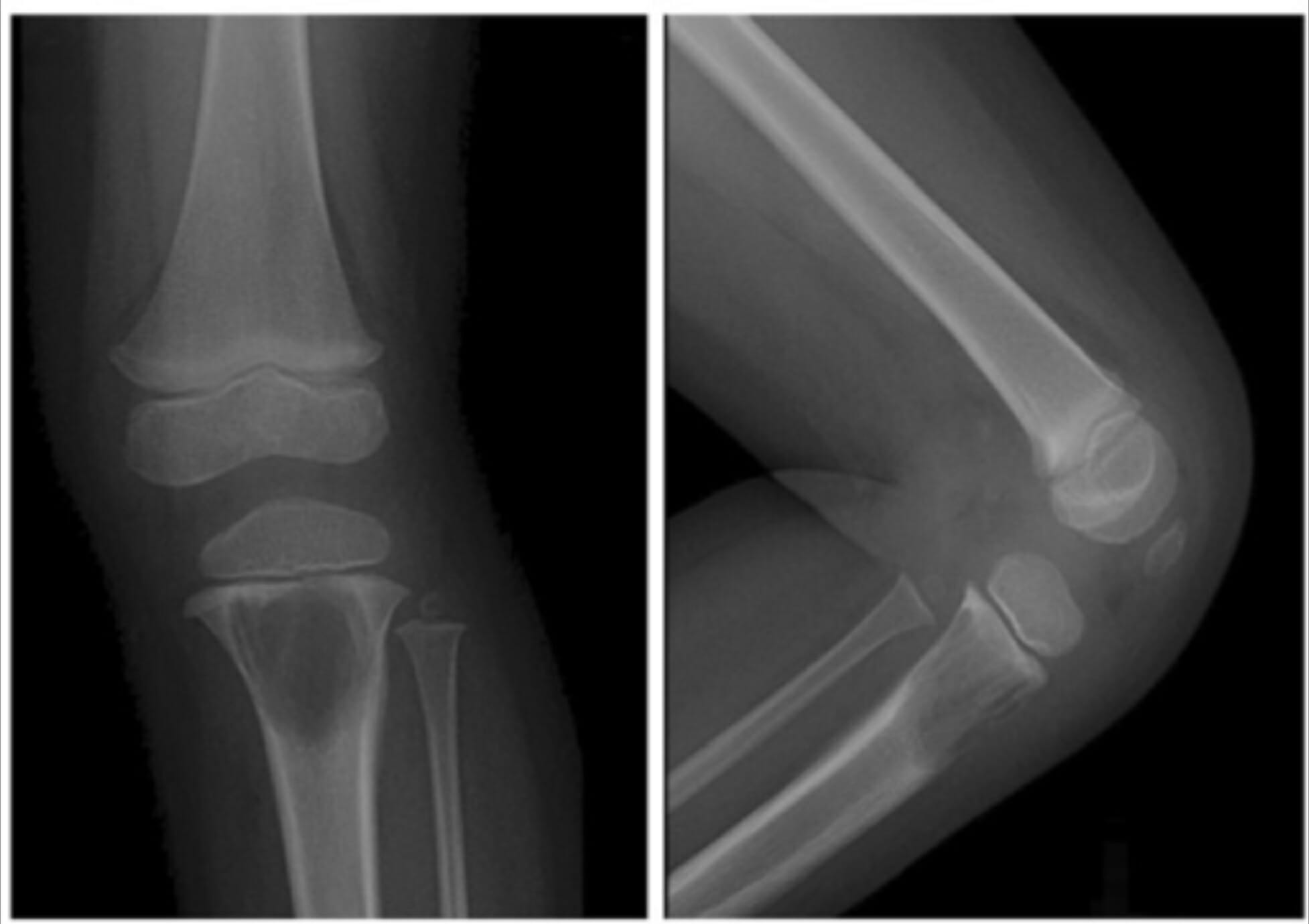
Radiografias do joelho esquerdo de uma criança a demonstrar uma grande lesão lítica na metáfise proximal da tíbia devido à histiocitose de células de Langerhans
Imagem: “A 2-year-old girl with left knee pain and a medullary lytic lesion in the proximal tibial metaphysis” por Department of Radiology, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan. Licença: CC BY 4.0
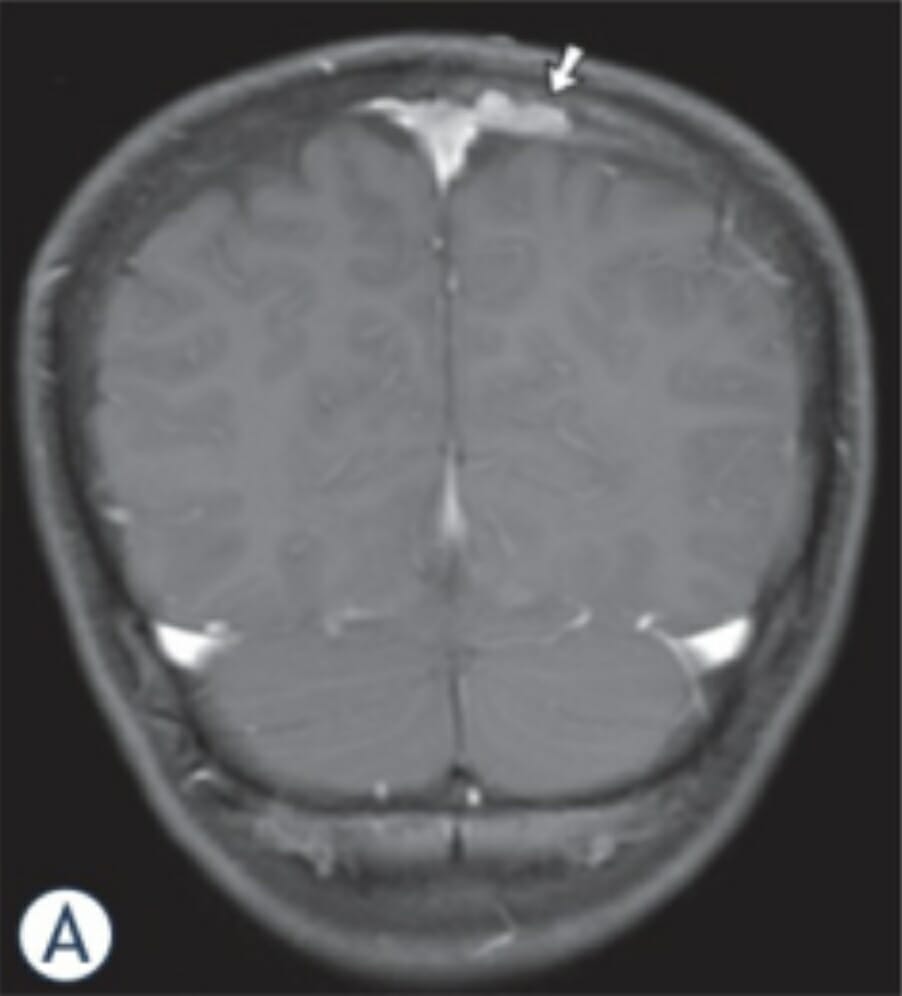
Imagens de RMN de um rapaz de 11 anos com HCL: a imagem coronal de RM com ponderação em T1 revela uma massa com realce ósseo (seta) combinada com envolvimento epidural e subdural ao longo do lado esquerdo do seio sagital superior.
Imagem: “MR-images in an 11-year-old boy with LCH” por Neuroradiology Department, Johann Wolfgang Goethe University, Frankfurt/Main, Germany. Licença: CC BY 3.0
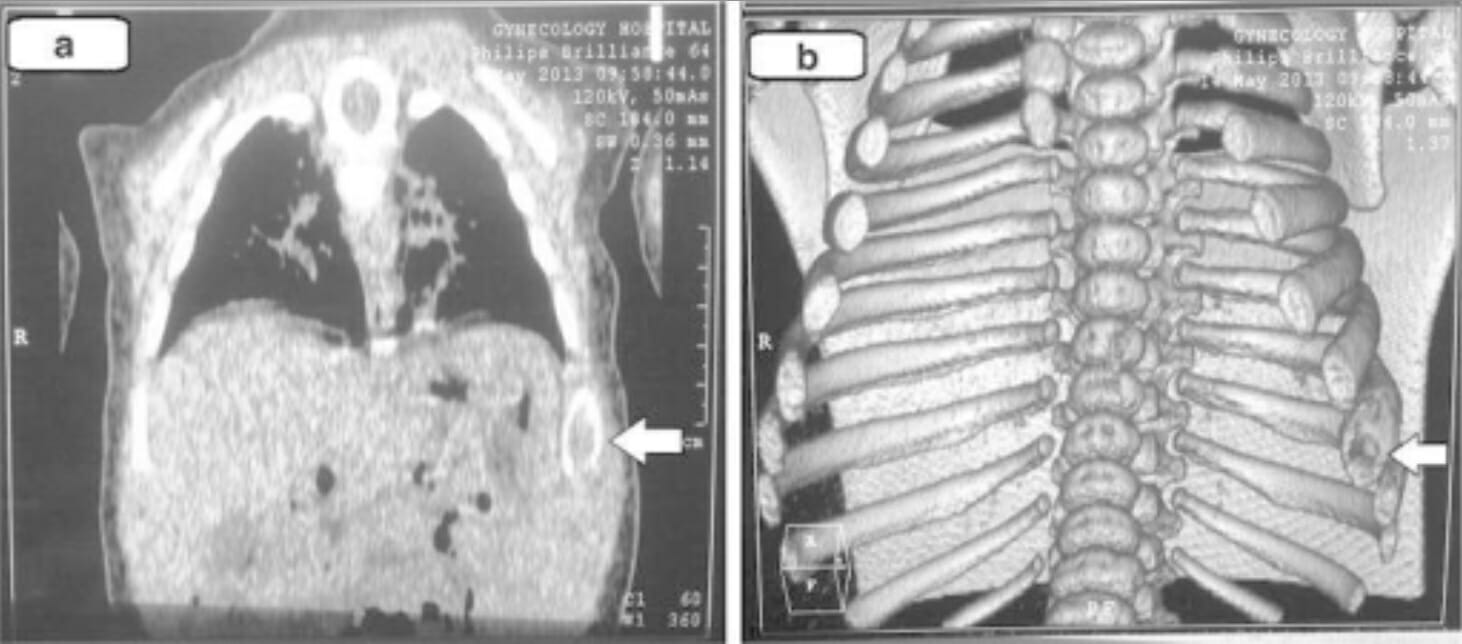
Achados da TC torácica (e imagens tridimensionais) na histiocitose de células de Langerhans: Estas imagens demonstram alterações líticas da 8ª costela esquerda (setas).
Imagem: “Chest computed tomography” por Unit of Pediatric Surgery, Al Diwaniya General Teaching Hospital, Al Qadisiya, Iraq. Licença: CC BY 4.0
Patologia
Exame de medula óssea (especialmente se hemograma anormal)
Biópsia da lesão/tumorTumorInflammation recomendada em todos os casos:
Exemplos:
Lesão óssea osteolítica
Lesão cutânea
Lavado broncoalveolar
Biópsia pulmonar
As características histopatológicas variam consoante o local da biópsia, mostrando:
Células de Langerhans (núcleos dobrados ou ranhurados, com citoplasma moderadamente abundante)
S100S100A family of highly acidic calcium-binding proteins found in large concentration in the brain and believed to be glial in origin. They are also found in other organs in the body. They have in common the ef-hand motif (ef hand motifs) found on a number of calcium binding proteins. The name of this family derives from the property of being soluble in a 100% saturated ammonium sulfate solution.Acoustic Neuroma
Genética:
Teste de mutação BRAF V600E (positivo em > 50%)
Considerar a sequenciação de nova geração para a via de sinalização MAPK-ERK
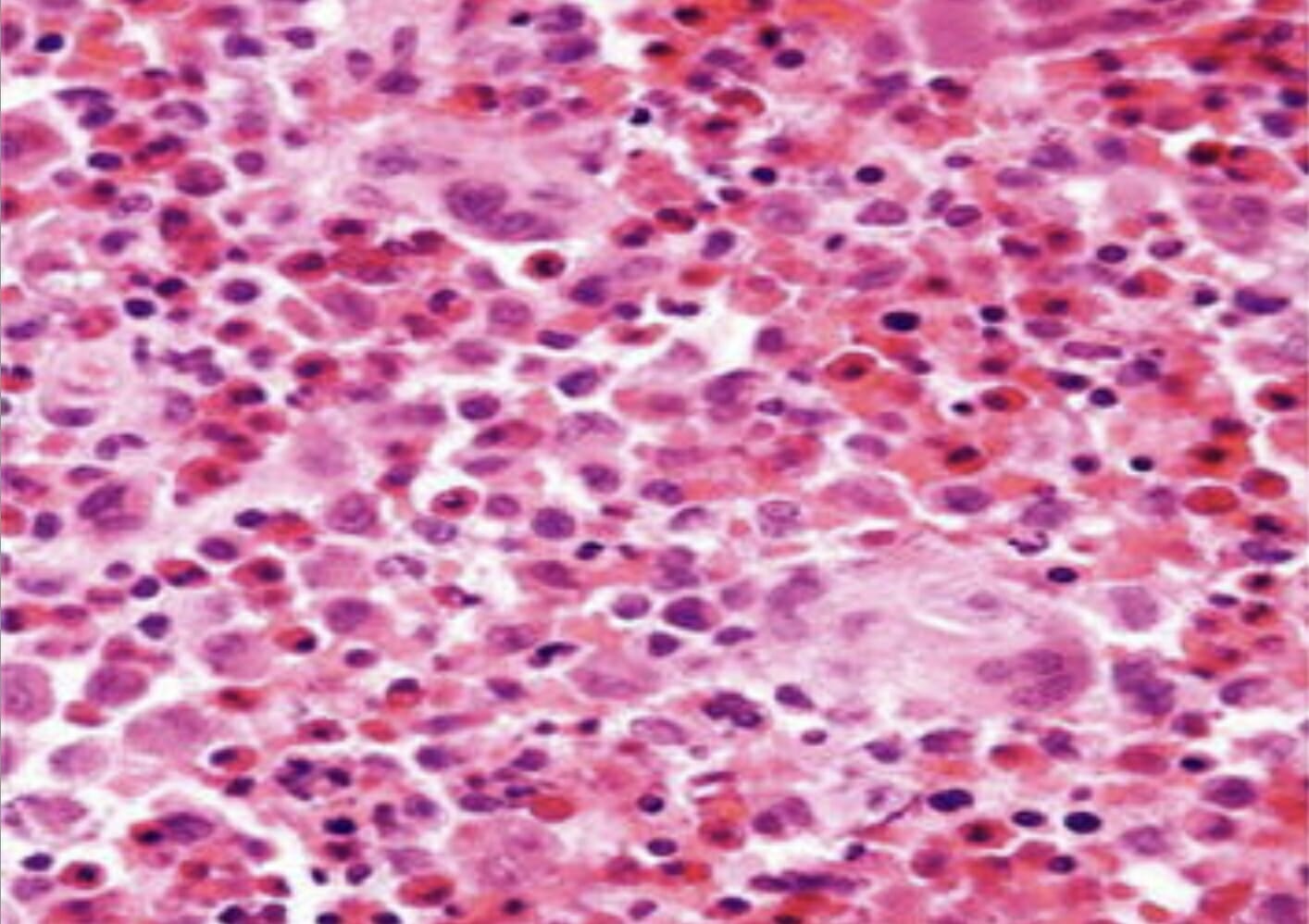
Biopsia histopatológica da pele (hematoxilina e eosina × 100) mostra agregados de células histiocíticas com um citoplasma eosinofílico e granular abundante
Imagem: “Skin biopsy histopathology” por Unit of Pediatric Surgery, Al Diwaniya General Teaching Hospital, Al Qadisiya, Iraq. Licença: CC BY 4.0
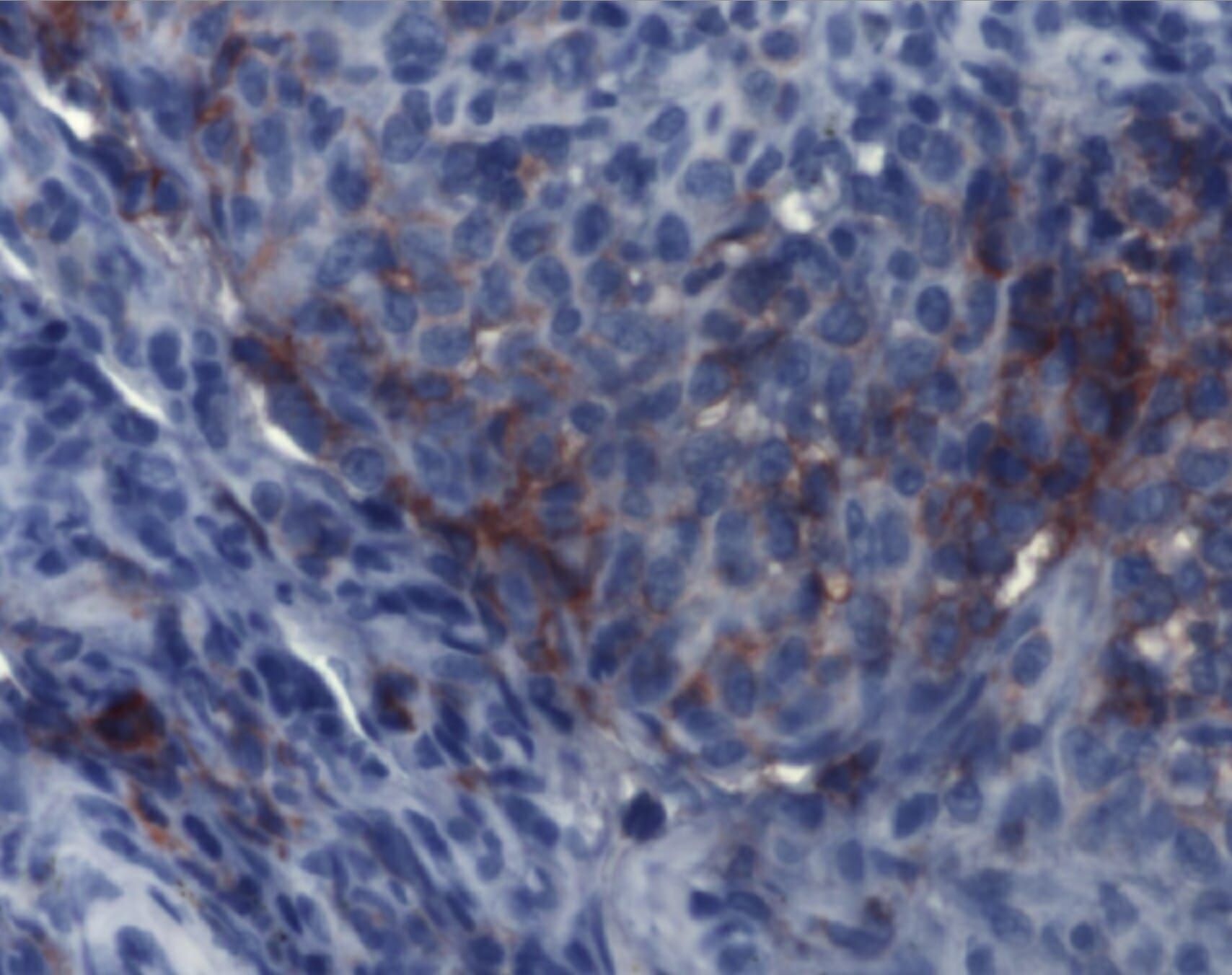
A imunohistoquímica demonstra achados consistentes com histiocitose de células de Langerhans. Os agregados de células de Langerhans são detetáveis por anticorpos monoclonais contra CD1a.
Imagem: “Immunohistochemistry of Langerhans cell histiocytosis” por Institute of Pathology and Neuropathology, University Hospital Essen, University of Duisburg-Essen, Germany. Licença: CC BY 2.0
Tratamento e Prognóstico
A escolha do tratamento da HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia baseia-se no tipo de HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia, apresentação clínica, órgãos envolvidos, grau de envolvimento e função dos órgãos. O tratamento é normalmente orientado por um oncologista (e eventualmente por outros especialistas).
Tratamento da HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar
Broncodilatadores inalados e/ou glicocorticoides se limitação reversível do fluxo de arARAortic regurgitation (AR) is a cardiac condition characterized by the backflow of blood from the aorta to the left ventricle during diastole. Aortic regurgitation is associated with an abnormal aortic valve and/or aortic root stemming from multiple causes, commonly rheumatic heart disease as well as congenital and degenerative valvular disorders. Aortic Regurgitation
Reabilitação pulmonar em doentes com dispneia de esforço
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar sintomática/progressiva:
Glicocorticoides sistémicos
Opções de quimioterapia:
Cladribina
Citarabina
Tratamento da HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia não pulmonar
Se assintomático ou sem disfunção de órgãos, pode ser considerada a observação.
Envolvimento multissistémicoem crianças:
Vimblastina
Prednisona
Envolvimento multissistémicoem adultos:
Opções de quimioterapia:
Citarabina
Cladribina
Terapêutica orientada para mutações genéticas conhecidas:
Inibidores de BRAF (e.g., vemurafenibe ou dabrafenibe)
Inibidores MEK (e.g., cobimetinibe)
Envolvimento de um único sistema:
Lesão óssea única (sem envolvimento do SNC):
Curetagem
Radioterapia:
Se lesão recorrente ou persistente
Não utilizado em crianças
A terapêutica com bisfosfonatos pode reduzir a dor.
Lesões ósseas múltiplas:
Terapêutica sistémica (quimioterapia e terapêuticas alvo semelhantes às utilizadas se envolvimento de múltiplos sistemas)
Tratamentos adjuvantes:
Tratamento cirúrgico
Radioterapia
Pele:
A HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia apenas cutânea pode regredir espontaneamente ou progredir para HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia multissistémica.
Doentes assintomáticos: observação
Sintomático ou com extenso envolvimento cutâneo:
Corticosteroides tópicos ou iperite
Metotrexato, que pode ser combinado com outros agentes imunossupressores (e.g., prednisolona, 6-mercaptopurina, hidroxiureia)
Lenalidomida
Talidomida
Prognóstico
O curso clínico e o prognóstico dos doentes com HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia são variáveis:
Envolvimento nodular único ou doença cutânea isolada
Ausência de lesões após 1 ano de seguimento
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar (em não fumador)
Associado a pior prognóstico:
Doenças multifocais ou doentes de alto risco
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia com mutações BRAF (maior probabilidade de recaída)
Envolvimento de órgãos de alto risco (e.g., SNC, medula óssea, fígado, baço)
A HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia crónica focal pode progredir para doença multifocalMultifocalRetinoblastoma ou disseminada.
Diagnóstico diferencial
Mieloma múltiplo: doença maligna na qual os plasmócitos proliferam anormalmente na medula óssea, causando o desvio das linhas celulares hematopoiéticas. A apresentação clínica pode incluir dor óssea, fraturas patológicas e sinais de insuficiência renal. As lesões ósseas osteolíticas podem ser visualizadas por imagem. As características de diagnóstico distintivas incluem a presença de proteína monoclonal no soro e na urina, a histologia (e.g., plasmócitos com citoplasma basofílico abundante) e o perfil imunofenotípico. O tratamento inclui corticosteroides, quimioterapia e/ou agentes imunomoduladores.
Doença de Erdheim-Chester (DEC): frequentemente uma doença do adulto, a DEC é uma doença histiocítica multissistémica. Os infiltrados xantogranulomatosos afetam ≥ 1 sistema orgânico (e.g., pele, pulmão, osso, cérebro, glândula hipofisária, retroperitoneu, sistema cardiovascular). Observa-se osteosclerose nos ossos longos e o diagnóstico é confirmado pelas características patológicas, como histiócitos espumosos e células gigantes de Touton (histiócitos multinucleados) num fundo de fibrose (sem expressão de CD1a).
Sarcoma de células de Langerhans: neoplasia de alto grau que se apresenta com envolvimento de um único órgão ou de múltiplos sistemas. A apresentação clínica é semelhante à da HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia, mas pode ser distinguida pela análise citológica. O sarcoma de células de Langerhans é caracterizado por citologia maligna (atipias celulares e mitoses atípicas).
Xantogranuloma juvenil: forma maisMAISAndrogen Insensitivity Syndrome comum de xantogranuloma não-HCL. O xantogranuloma juvenil afeta predominantemente as crianças e apresenta-se clinicamente com lesões cutâneas solitárias ou múltiplas. A dermoscopia ajuda no diagnóstico. A doença pode resolver-se espontaneamente ou pode exigir excisões cutâneas curativas.
Doença de Rosai-Dorfman: proliferação anormal e acumulação de histiócitos nos gânglios linfáticos, com rara ocorrência de envolvimento extranodal. Os indivíduos podem apresentar linfadenopatia (cervical), febre, palidez, perda de peso, mal-estar e rinite crónica. O diagnóstico envolve o exame histopatológico (negativo para CD1a e CD207). A remissão espontânea pode ocorrer. Os tratamentos incluem terapêutica de suporte, administração de corticosteroides, alfa-interferão ou quimioterapia.
Emile, J. F., Abla, O., Fraitag, S., Horne, A., Haroche, J., Donadieu, J., Requena-Caballero, L., Jordan, M. B., Abdel-Wahab, O., Allen, C. E., Charlotte, F., Diamond, E. L., Egeler, R. M., Fischer, A., Herrera, J. G., Henter, J. I., Janku, F., Merad, M., Picarsic, J., Rodriguez-Galindo, C., … Histiocyte Society. (2016). Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood, 127(22), 2672–2681. https://doi.org/10.1182/blood-2016-01-690636
Goyal, G., Young, J. R., Koster, M. J., Tobin, W. O., Vassallo, R., Ryu, J. H., Davidge-Pitts, C. J., Hurtado, M. D., Ravindran, A., Sartori Valinotti, J. C., Bennani, N. N., Shah, M. V., Rech, K. L., Go, R. S., Mayo Clinic Histiocytosis Working Group. (2019). The Mayo Clinic Histiocytosis Working Group consensus statement for the diagnosis and evaluation of adult patients with histiocytic neoplasms: Erdheim-Chester disease, Langerhans cell histiocytosis, and Rosai-Dorfman disease. Mayo Clinic Proceedings, 94(10), 2054–2071. https://doi.org/10.1016/j.mayocp.2019.02.023
Allen, C. E., Merad, M., McClain, K. L. (2018). Langerhans-cell histiocytosis. New England Journal of Medicine, 379(9), 856–868. https://doi.org/10.1056/NEJMra1607548
Jezierska, M., Stefanowicz, J., Romanowicz, G., Kosiak, W., Lange, M. (2018). Langerhans cell histiocytosis in children—a disease with many faces: recent advances in pathogenesis, diagnostic examinations and treatment. Advances in Dermatology and Allergology, 35(1), 6–17. https://doi.org/10.5114/pdia.2017.67095
Kumar, V., Abbas, A., Aster, J., Turner, J. (2021). Diseases of the white blood cells, lymph nodes, spleen and thyroid. In Robbins and Cotran Pathologic Basis of Disease (10th ed., pp. 627–628). Elsevier.
Go, R. S., et al. (2021). Histiocytic neoplasms, version 2.2021, NCCN clinical practice guidelines in oncology. JNCCN, 19(11), 1277–1303. https://doi.org/10.6004/jnccn.2021.0053
Vassallo, R., Harari, S., Tazi, A. (2017). Current understanding and management of pulmonary Langerhans cell histiocytosis. Thorax, 72(10), 937–945. https://doi.org/10.1136/thoraxjnl-2017-210125
¡Crea tu cuenta gratis o inicia una sesión para seguir leyendo!
A Lecturio Medical complementa o teu estudo através de métodos de ensino baseados em evidência, vídeos de palestras, perguntas e muito mais – tudo combinado num só lugar e fácil de usar.
User Reviews
Details
×
Obtenha Premium para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características
Obtenha Premium para ver todos os vídeos
Verifique agora o seu e-mail para obter um teste gratuito.
Crie uma conta gratuita para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características - incluindo o Qbank de Lecturio com perguntas actualizadas ao estilo do board-.