


A glomerulonefrite membranoproliferativa (GNMP) também é conhecida como glomerulonefrite mesangiocapilar. A glomerulonefrite membranoproliferativa é um padrão de lesão glomerular caracterizada por hipercelularidade mesangial, proliferação endocapilar e espessamento da membrana basal glomerular (formação de contorno duplo). As alterações são causadas pela deposição de Igs, de fatores do complemento, ou ambos, no mesângio glomerular e ao longo das paredes capilares glomerulares. As variantes patogénicas incluem a mediada por imunocomplexos/Ig monoclonal (e.g., de infeções, doenças autoimunes) e GNMP mediada por complemento. Em casos raros, a GNMP não está associada às Igs e ao sistema complemento, como no caso de lesão endotelial. Tendo múltiplas etiologias, a apresentação e o curso clínico variam. As características de apresentação podem ser a proteinúria e a hematúria assintomática, a síndrome nefrótica, a síndrome nefrítica ou a insuficiência renal crónica. O diagnóstico definitivo requer biópsia renal, embora exames laboratoriais e de imagem adicionais possam apontar para a doença associada. O tratamento é focado na causa subjacente. Os esteroides, imunossupressores e transplante renal estão entre as modalidades de tratamento frequentemente utilizadas.
Last updated: Dec 15, 2025
A glomerulonefrite membranoproliferativa (GNMP) é uma lesão glomerular caracterizada por espessamento da membrana basal glomerular (MBG) (“membrano-”) e aumento da celularidade endocapilar e mesangial (“proliferativa”).
Tradicionalmente, a GNMP era classificada com base nos achados da microscopia eletrónica (classificação antiga):
Uma classificação diferente, baseada no processo patogenético, ajuda a orientar a etiologia ou doença subjacente, direcionando o tratamento.
| GNMP mediada por imunocomplexos/monoclonal | GNMP mediada por complemento | GNMP sem Ig ou complemento | |
|---|---|---|---|
| LM | “Linha de comboio” (duplo contorno) da membrana basal | ||
| IM |
|
Complemento positivo e sem (ou mínima) coloração para Ig | Sem coloração de Ig ou complemento |
| EM | Depósitos subendoteliais e mesangiais (em algumas doenças autoimunes, + depósitos subepiteliais) |
|
Sem depósitos eletrodensos ao longo das paredes capilares |
| Diagnóstico Diferencial |
|
|
Lesão endotelial, que pode ser de:
|
| Etiologia | Achados microscópicos | |
|---|---|---|
| GNMP mediada por imunocomplexos/monoclonal | Hepatite B ou C (ou outras infeções virais) |
|
| Gamopatia monoclonal | Cadeias leves kappa OR lambda | |
| Doenças autoimunes | “Padrão full-house”:
|
|
| GNMP mediada por complemento | Glomerulopatia C3 |
|
| Glomerulopatia C4 |
|
|
| GNMP não associada ao complemento ou deposição de Ig | Microangiopatias (frequentemente associadas a lesão endotelial) | Sem observação de deposição significativa de Ig ou complemento |
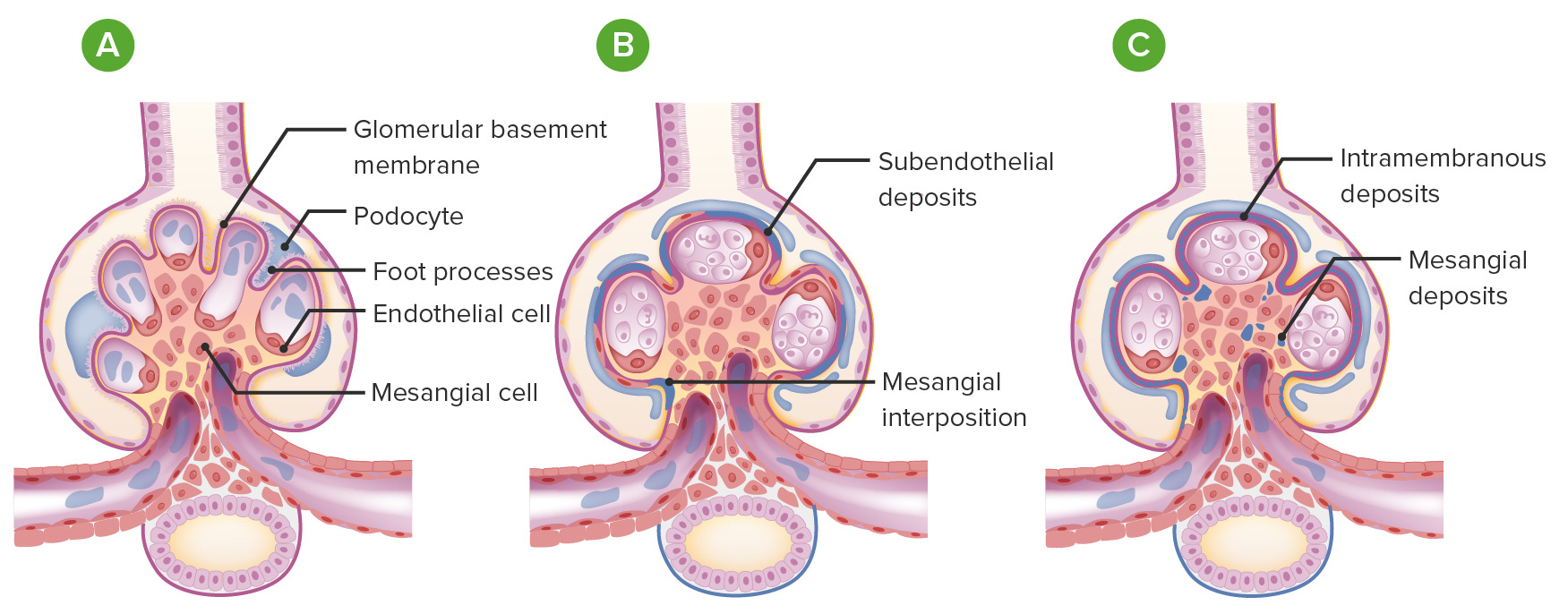
Glomerulonefrite membranoproliferativa (GNMP) vs glomérulos normais:
A: glomérulo normal (com ansas capilares abertas, ≤ 3 núcleos em cada área mesangial, processos podocitários intactos e sem depósitos ou proliferação)
B: GNMP: Os glomérulos tornam-se lobulados com proliferação endocapilar e a membrana basal glomerular tem uma aparência dividida (por depósitos subendoteliais e interposição mesangial).
C: GNMP com depósitos mesangiais e depósitos intramembranosos (como visto na doença de depósitos densos)
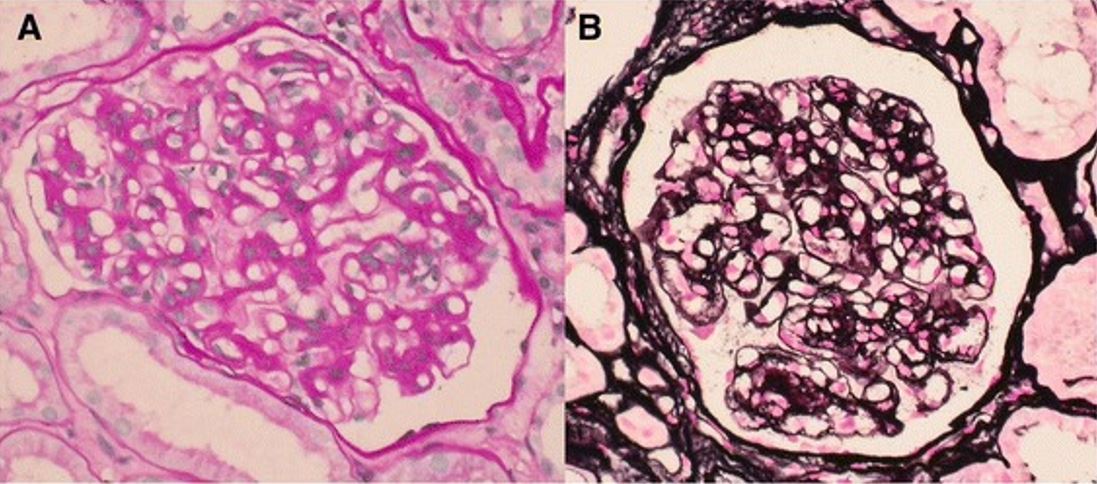
Características ao microscópio ótico da glomerulonefrite membranoproliferativa
A: O mesangio está expandido e as paredes dos capilares glomerulares aparecem espessadas (ácido periódico-Schiff).
B: As paredes capilares glomerulares apresentam contornos duplos, espessados e segmentares (prata metenamina).
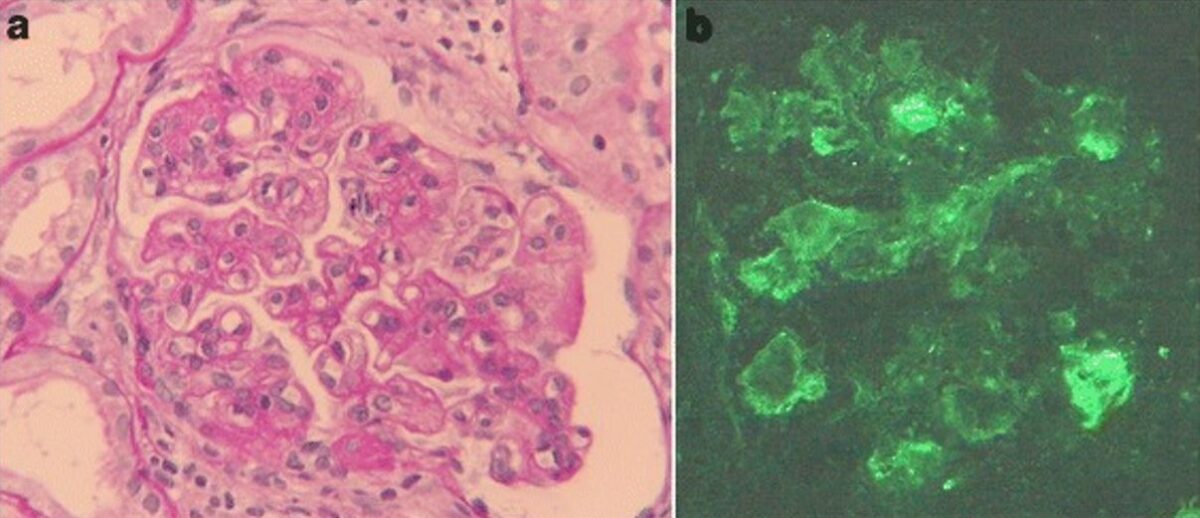
Glomerulonefrite membranoproliferativa associada à hepatite C
a: achados na microscopia ótica da biópsia renal: a coloração com ácido periódico de Schiff revela hipercelularidade mesangial, acentuação lobular e contorno duplo da membrana basal (ampliação original, 400×).
b: A coloração de imunofluorescência de IgM é positiva ao longo da ansa capilar (ampliação original, 400×).
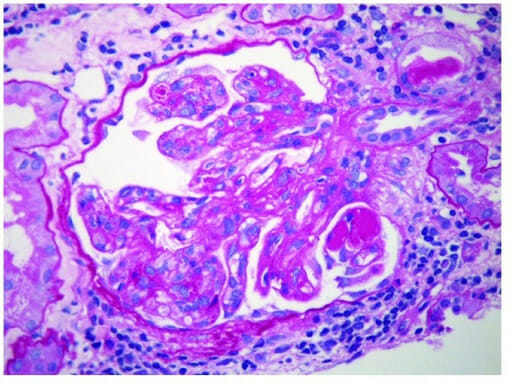
Glomerulonefrite membranoproliferativa:
Proliferação endocapilar com extensos depósitos subendoteliais ao longo das paredes capilares glomerulares. Também estão presentes depósitos mesangiais (microscopia eletrónica).
A investigação adicional é indicada quando a biópsia mostra alterações consistentes com:
