


A doença de Von Hippel-Lindau (VHL) é uma condição genética autossómica dominante resultante de uma deleção ou mutação no gene VHL. Indivíduos diagnosticados com doença de VHL apresentam tumores e cistos em várias partes do corpo e podem apresentar hemangioblastomas, carcinoma de células renais (CCR), feocromocitoma, tumores do saco endolinfático do ouvido médio, tumores pancreáticos e cistoadenomas papilares do epidídimo ou do ligamento largo. O diagnóstico é baseado em testes genéticos, na avaliação laboratorial do BUN, avaliação laboratorial da presença de catecolaminas no sangue ou na urina, exame fundoscópico do olho para detetar hemangioblastomas da retina e TAC/RMN para detetar quaisquer outros tumores. O tratamento da doença inclui a remoção cirúrgica dos tumores.
Last updated: Dec 15, 2025
A doença de Von Hippel-Lindau (VHL) é uma doença autossómica dominante caracterizada por hemangioblastomas Hemangioblastomas A benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia. Von Hippel-Lindau Disease da retina Retina The ten-layered nervous tissue membrane of the eye. It is continuous with the optic nerve and receives images of external objects and transmits visual impulses to the brain. Its outer surface is in contact with the choroid and the inner surface with the vitreous body. The outermost layer is pigmented, whereas the inner nine layers are transparent. Eye: Anatomy e do SNC; cistos envolvendo os rins, pâncreas e epidídimo; carcinoma de células renais (CCR); feocromocitomas; e tumores de células das ilhotas pancreáticas.
A apresentação varia dependendo do tamanho e localização do tumor Tumor Inflammation. A história familiar é uma informação fundamental, pois a maioria dos casos é herdado. O exame físico geralmente não é revelador, com exceção de indivíduos afetados que apresentam alterações neurológicas no contexto de hemangioblastomas Hemangioblastomas A benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia. Von Hippel-Lindau Disease.
HIPPEL :
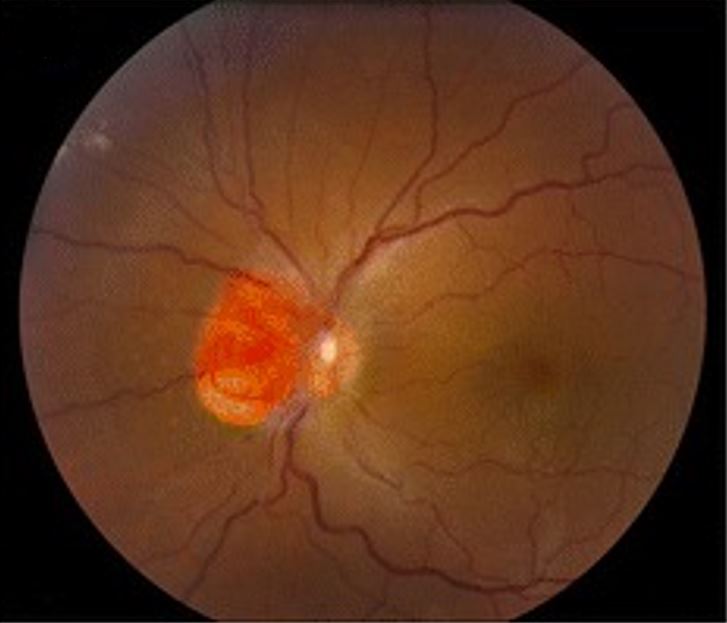
Hemangioblastoma a envolver o nervo ótico, como visto na doença de von Hippel-Lindau
Imagem: “Clinical photographs of the eye from the patient with von Hippel-Lindau disease” por Chen S, Chew EY, Chan CC. Licença: CC BY 4.0, editado por Lecturio.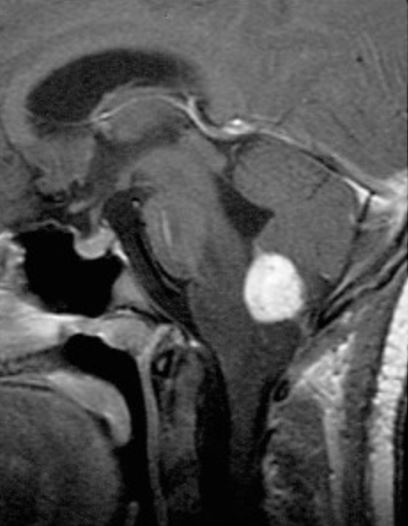
RMN sagital, contrastada, ponderada em T1 a mostrar um hemangioblastoma medular com contraste
Imagem: “Radiographic images of hemangioblastomas” por Schunemann V, Huntoon K, Lonser RR. Licença: CC BY 4.0, editado por Lecturio.O tratamento da doença de VHL é adaptado dependendo da localização e tamanho das lesões, sintomas do indivíduo e extensão da doença.
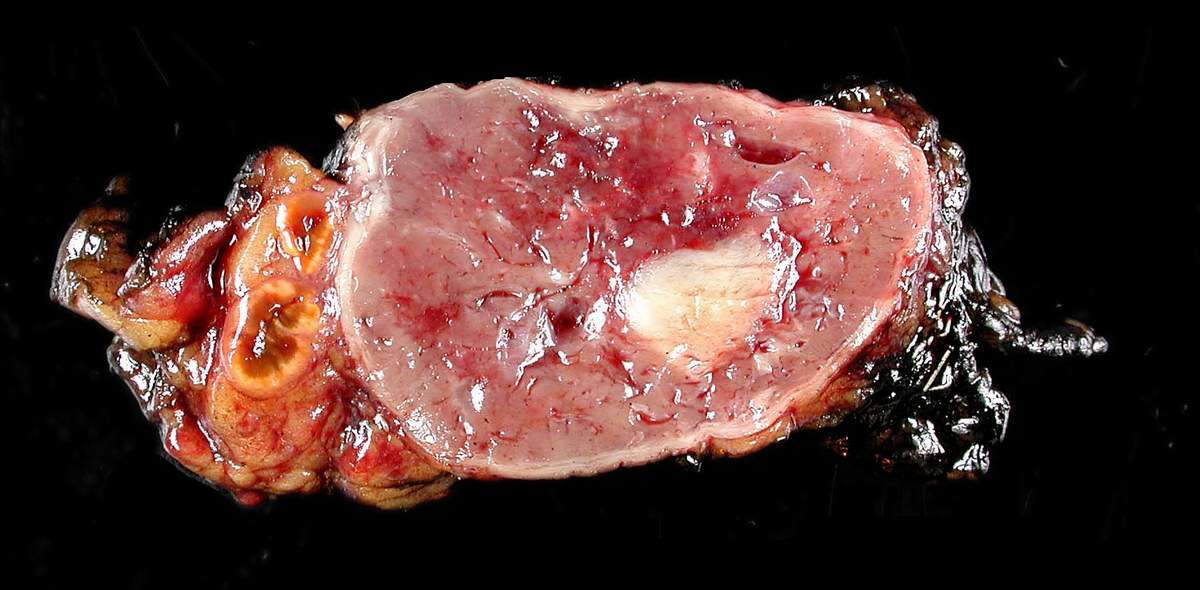
Feocromocitoma removido cirurgicamente
Imagem: “Adrenal paraganglioma clinical Pheochromocytoma” por Michael Feldman. Licença: CC BY 2.0