As anomalias da córnea são condições nas quais a estrutura ou a função da córnea está comprometida devido a várias patologias congénitas ou adquiridas. As anomalias da córnea são classificadas com base em alterações do tamanho, transparência, anomalias ectáticas, distrofias da córnea e patologias adquiridas. As alterações patológicas podem resultar em opacidade ou turvação da córnea com consequente redução da acuidade visual. As anomalias da córnea são geralmente diagnosticadas com base em achados clínicos. A abordagem inclui a correção de erros refrativos e o tratamento das condições subjacentes.
A curvatura da córnea é > ao restante globo ocular.
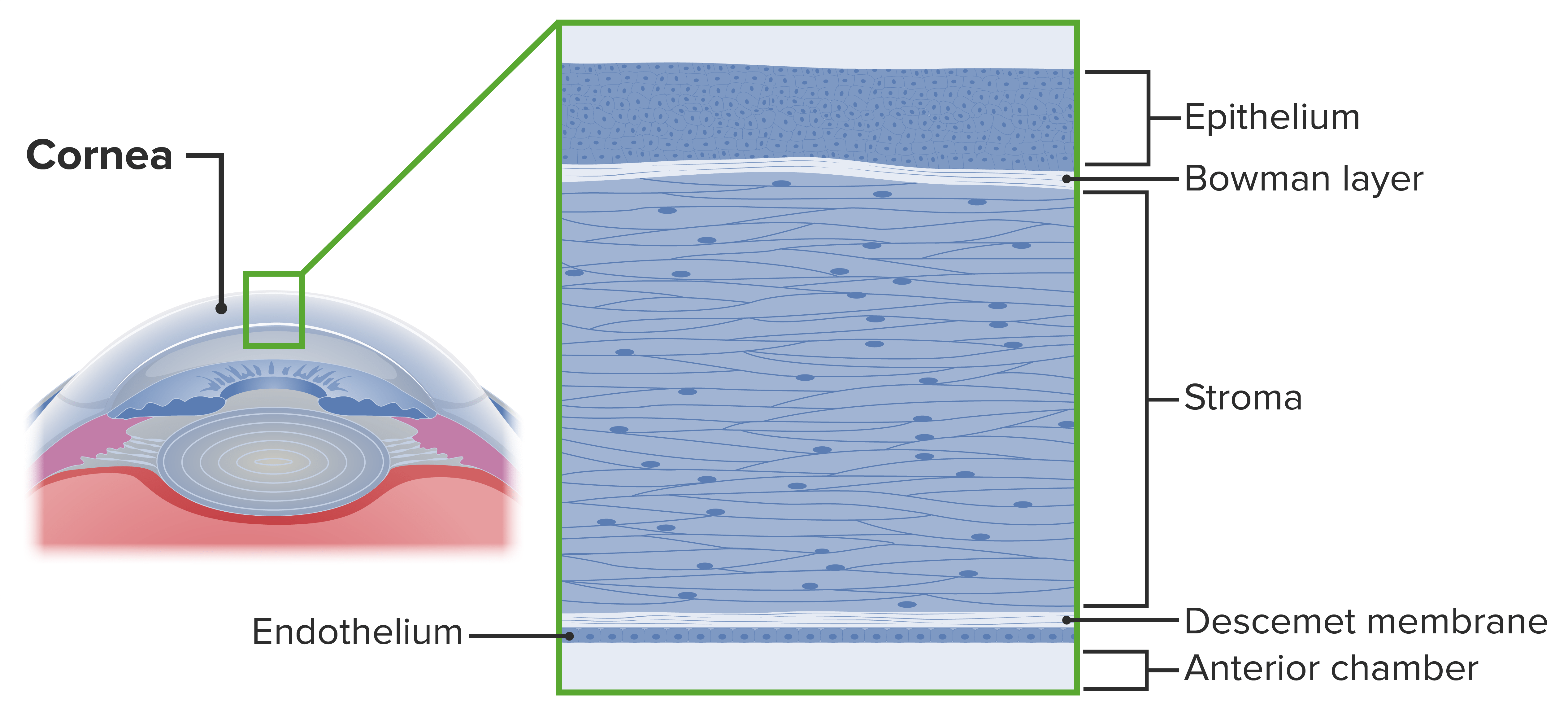
5 camadas histológicas:
Epitélio
Membrana de Bowman
Estroma
Membrana de Descemet
Endotélio
Classificação das anomalias da córnea
Criptoftalmia
Anomalias do tamanho:
Microcórnea
Megalocórnea
Anomalias ectáticas:
Queratocone
Queratoglobo
Anomalias da transparência:
Anomalia de Peters
Esclerocórnea
Distrofias da córnea:
Distrofia estromal congénita da córnea
Distrofia endotelial congénita da córnea
Patologias adquiridas:
Queloides da córnea
Queratopatia em banda
Camadas da córnea
Imagem por Lecturio.
Criptoftalmia
A criptoftalmia é uma doença congénita autossómica recessiva associada a anomalias sistémicas, sendo caracterizada por uma continuidade ininterrupta da pele que se estende desde a testa até à região malar.
Epidemiologia
A incidência de criptoftalmia é desconhecida.
Pode ocorrer isoladamente
Também pode ocorrer associada à síndrome de Fraser, uma doença autossómica recessiva, caracterizada por:
As pálpebras são substituídas por uma camada de pele que se estende desde a testa até a região malar.
Ausência ou fraco desenvolvimento da sobrancelha
A córnea está ausente ou fundida com a pele.
Parcial = resulta de um desenvolvimento anormal da prega palpebral
Abrange cerca de 20% dos casos
Estão presentes pálpebras rudimentares com um pequeno saco conjuntival lateralmente.
O globo ocular é pequeno e, na sua maioria, é recoberto por pele.
Abortivo = A área da prega palpebral que deriva do processo frontonasal não se desenvolve, enquanto a área que deriva do processo maxilar desenvolve-se normalmente.
A pálpebra superior anormal cobre e adere até 75% da córnea superior.
Sem ponto ou fórnix conjuntival superior
Pálpebra inferior normal
Tratamento
O tratamento inclui a reconstrução cirúrgica das pálpebras para permitir o desenvolvimento visual. Não existe uma abordagem principal e as cirurgias necessárias variam conforme a gravidade das anomalias e o desenvolvimento da órbita.
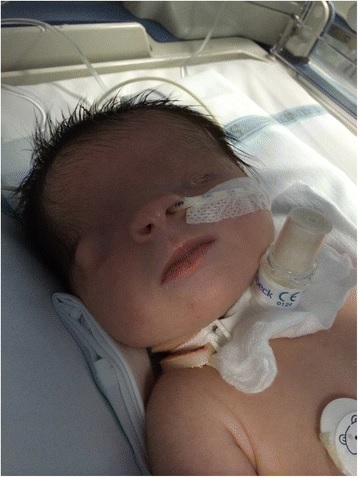
Criptoftalmia bilateral
Imagem: “Bilateral cryptophthalmos with microphthalmos into left ocular globe and abnormal right ocular globe in a female infant with Fraser syndrome” por De Bernardo et al. Licença: CC BY 4.0
Anomalias do Tamanho da Córnea
Microcórnea
O diâmetro horizontal normal da córnea ao nascimento é de 10 mm. O tamanho adulto é cerca de 11,7 mm e é normalmente atingido por volta dos 2 anos de idade.
Descrição geral:
A microcórnea caracteriza-se por um diâmetro da córnea < 10 mm.
Pode ser unilateral ou bilateral
Pode ocorrer como uma anomalia isolada
Pode estar associada a:
Microftalmia: globo ocular anormalmente pequeno
Nanoftalmia: olho anormalmente pequeno com cristalino de tamanho normal
Apresentação clínica:
Geralmente associada a miopia e microftalmia
Pode apresentar-se como microcórnea isolada ou microftalmia anterior relativa
Os indivíduos geralmente desenvolvem glaucomaGlaucomaGlaucoma is an optic neuropathy characterized by typical visual field defects and optic nerve atrophy seen as optic disc cupping on examination. The acute form of glaucoma is a medical emergency. Glaucoma is often, but not always, caused by increased intraocular pressure (IOP). Glaucoma na 4.ª década de vida.
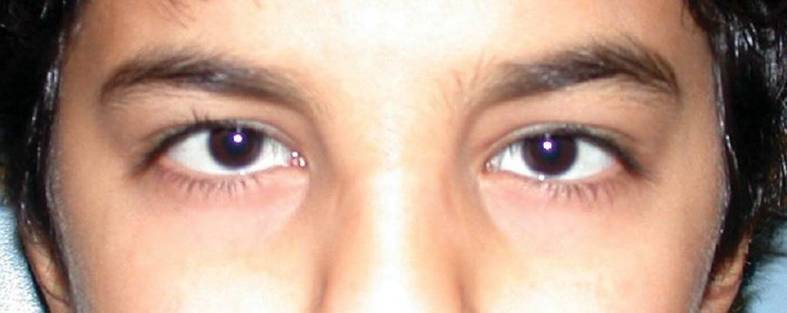
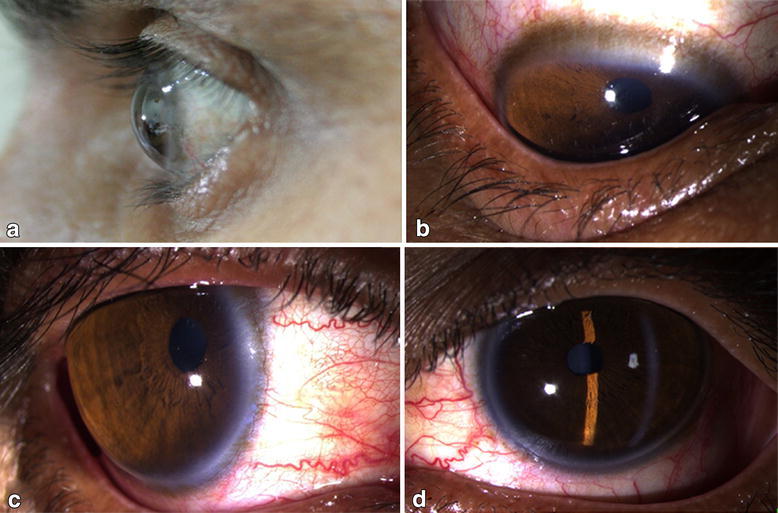
Criança com microcórnea (diâmetros < 10 mm)
Imagem: “At 12 years of age, the child had horizontal corneal diameters of 10 mm but otherwise unremarkable anterior segments by slit-lamp examination” por Hazin R et al. Licença: CC BY 2.0
Megalocórnea
A megalocórnea caracteriza-se por um aumento da córnea bilateral, não progressivo, com um diâmetro horizontal da córnea > 12 mm ao nascimento e 13 mm aos 2 anos de idade.
Descrição geral:
Normalmente, córnea transparente com espessura e visão normais
Transmissão hereditária como traço recessivo associado ao X
Associação a patologias sistémicas:
Síndrome de Marfan
Síndrome de Ehlers-Danlos
Síndrome de Down
Fisiopatologia:
Durante o desenvolvimento da córnea, um defeito na formação do copo ótico permite que este cresça em demasia.
Considerado um crescimento excessivo primário da córnea
Densidade normal de células endoteliais.
Apresentação clínica:
Apresenta-se após os 12 meses de idade, quando a córnea se encontra totalmente desenvolvida
Córneas simétricas, em forma de cúpula com > 13 mm de diâmetro
A diminuição da acuidade visual é incomum.
O astigmatismo é comum.
Megalocórnea em criança com diâmetro da córnea > 14 mm
Imagem: “Megalocornea in a child with a corneal diameter greater than 14 mm” por Khan AO et al. Licença: CC BY 3.0, recortado por Lecturio.
Anomalias Ectáticas da Córnea
Queratocone
Queratocone é o resultado da diminuição da espessura da córnea junto ao centro, com consequente aumento da sua inclinação, assumindo uma forma cónica.
Descrição geral:
Não inflamatório
Doença progressiva
Causada por uma fragilidade congénita da córnea
Pode ser secundária a queratoconjuntivite vírica ou trauma
Apresentação clínica:
Apresenta-se na puberdade ou no início da idade adulta
O défice visual progride até à 4.ª década de vida.
Dificuldades na correção visual para miopia e astigmatismo
Achados com a doença avançada:
Sinal de Munson: deformação em forma de v da pálpebra inferior no olhar para baixo (ver imagem)
Estrias de Vogt: linhas finas no estroma profundo, patognomónicas para queratocone
Anel de Fleischer: deposição de hemossiderina na base do cone
Hidrópsia aguda: ruturas que se desenvolvem na membrana de Descemet, tornando o estroma subitamente edemaciado e opaco.
Pode apresentar-se com fotofobia e diminuição súbita e dolorosa da acuidade visual
Os tratamentos incluem adesivos oculares e lentes de contacto com intuito curativo.
Também são utilizados hiperosmóticos tópicos, cicloplégicos e corticóides para diminuir o edemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema, a dor e a inflamação.
Diagnóstico:
Exame com lâmpada de fenda: a córnea pode parecer normal.
Queratometria: pode mostrar distorção
Topografia da córnea: pode auxiliar no diagnóstico e na avaliação da progressão da doença
Tratamento:
Crosslinking do colagénio corneano: um procedimento que utiliza gotas de riboflavina, luz ultravioleta e um fotossensibilizador para fortalecer as ligações na córnea e medir a progressão.
Óculos: para corrigir o astigmatismo miópico, desde que o paciente consiga tolerar
Lentes de contacto:
Lentes rígidas permeáveis a gases neutralizam a irregularidade da córnea, mas podem tornar-se intoleráveis.
Também podem ser utilizadas lentes esclerais, que permanecem na esclera sem tocar na córnea.
Intervenções cirúrgicas:
Queratoplastia: necessária em 10 %–15% dos pacientes
Segmentos de anel corneano intraestromal
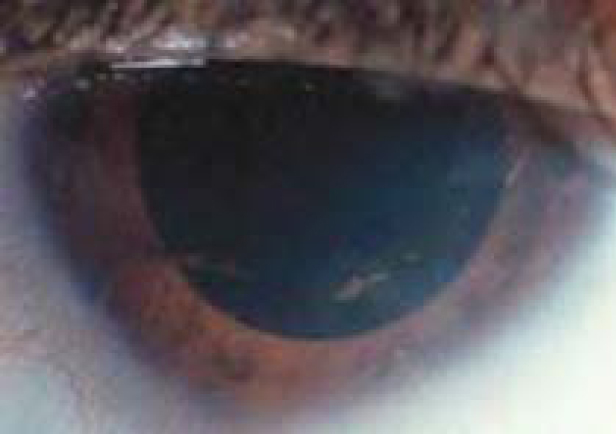
Sinal de Munson em paciente com queratocone
Imagem: “Münson’s sign in a keratoconus patient which appears as bulging of the lower lid during downgaze” por BioMed Research International/Mariam Lotfy Khaled et al. Licença: CC BY 4.0
Queratoglobo
Queratoglobo é uma protrusão hemisférica de toda a córnea com diminuição da espessura limbo a limbo da córnea.
Descrição geral:
Se congénita, manifesta-se bilateralmente.
Formas adquiridas também podem ocorrer devido a:
Queratoconjuntivite
Blefarite marginal crónica
Inflamação orbitária idiopática
Origem não inflamatória
De natureza progressiva
Forte associação com a síndrome de Ehlers-Danlos tipo VI
Geralmente associada a defeitos na síntese ou degradação do colagénio
Apresentação clínica:
Córneas transparentes, a menos que existam episódios de hidrópsia e cicatrizes
Alta miopia com astigmatismo irregular
Perfurações da córnea devido à diminuição excessiva da espessura
Tratamento:
Correção refrativa da alta miopia
Os óculos de proteção reduzem a incidência de perfurações da córnea.
Lentes esclerais personalizadas
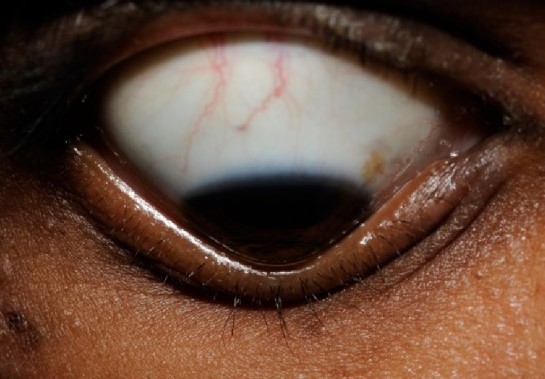
a: diminuição difusa da espessura da córnea com protrusão externa do globo b: deformação da pálpebra inferior c e d: limbo alongado com câmara anterior transparente
Imagem: “Clinical picture of the patient showing diffuse thinning of cornea with outward globular protrusion” por Gupta N et al. Licença: CC BY 4.0
A anomalia de Peters é uma forma congénita de disgenesia do segmento anterior onde a clivagem anormal da câmara anterior resulta num defeito central no endotélio da córnea e causa leucoma (placa esbranquiçada).
Descrição geral:
Loci de múltiplos genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure:
PAX6
PITX2
FOXC1
CYP1B1
Pode ocorrer esporadicamente, mas formas de transmissão dominante e recessiva são comuns
A associação a condições sistémicas inclui:
Trissomia 13
Trissomia 15
Deleção parcial do braço do cromossoma 11q
A incidência é desconhecida: uma parte das opacidades congénitas da córnea estão presentes em 3–6 indivíduos por 100.000
Patogénese: devido a uma interrupção na migração ou separação da crista neural que ocorre na 7.ª semana de gestação
Apresentação clínica e diagnóstico:
Está sempre presente opacidade da córnea central, para central ou completa.
São frequentemente observadas sinéquias da íris através da câmara anterior até a superfície posterior da córnea.
Diagnóstico: baseado em achados clínicos e confirmado por ecografia
Tratamento:
Monitorização regularRegularInsulin para glaucomaGlaucomaGlaucoma is an optic neuropathy characterized by typical visual field defects and optic nerve atrophy seen as optic disc cupping on examination. The acute form of glaucoma is a medical emergency. Glaucoma is often, but not always, caused by increased intraocular pressure (IOP). Glaucoma
Cirurgia: queratoplastia penetrante de espessura total com lensectomia em pacientes com catarata
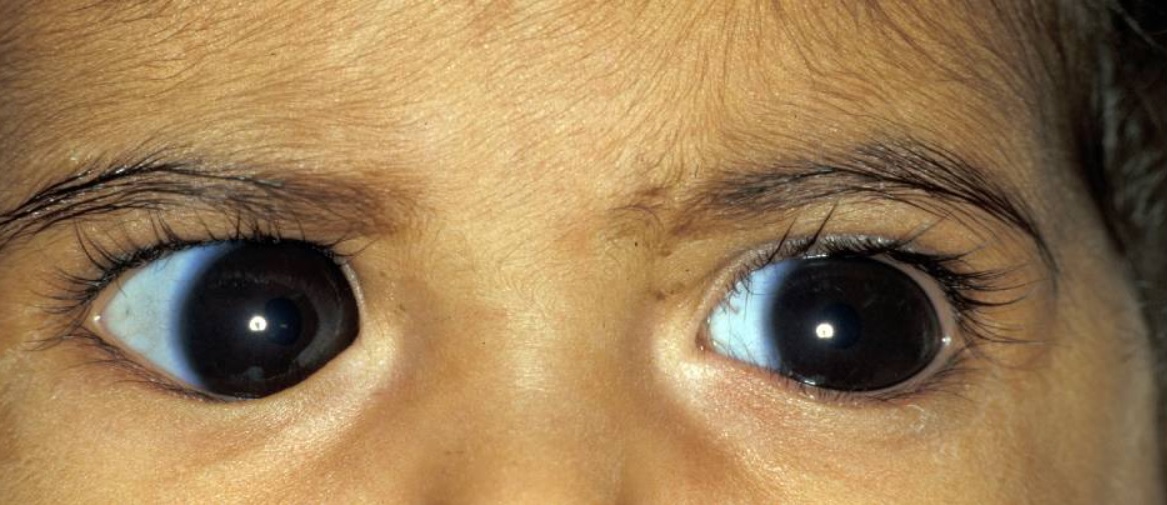
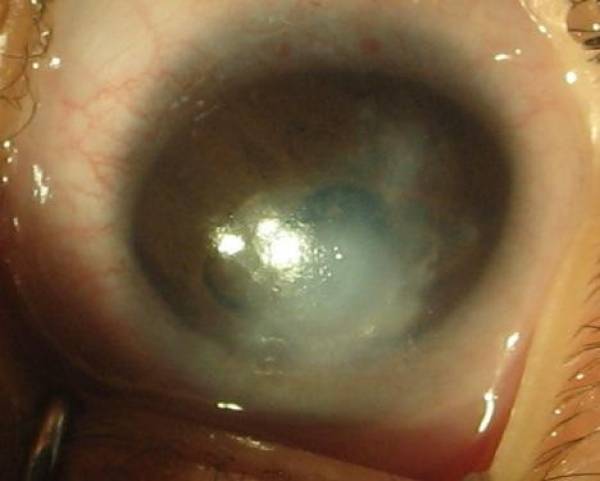
Opacidade da córnea em paciente com anomalia de Peters
Imagem: “Photograph of left eye” por Tuli N et al. Licença: CC BY 2.0
Esclerocórnea
Na esclerocórnea, os limites da córnea e da esclera são indistintos, resultando numa anomalia congénita da transparência. São observadas fibras de colagénio dispostas irregularmente.
Descrição geral:
Estão frequentemente presentes outras anomalias oculares, como a catarata ou o colobomaColobomaCongenital anomaly in which some of the structures of the eye are absent due to incomplete fusion of the fetal intraocular fissure during gestation.Esophageal Atresia and Tracheoesophageal Fistula.
Não progressivo
Não inflamatório
Pode afetar toda a córnea ou estar limitado a uma parte da mesma:
Esclerocórnea do tipo periférico: a área afetada é vascularizada com vasos esclerais superficiais.
Esclerocórnea total: toda a córnea é opaca e vascularizada.
O desenvolvimento da córnea ocorre durante a 7.ª e 8.ª semanas de gestação.
A falha na diferenciação das células mesenquimatosas em células da córnea e da esclera permite que a curvatura da córnea exceda a da esclera.
Apresentação clínica:
Esclerocórnea parcial: presença de borda córnea periférica, branca, vascularizada com 1–2 mm que se funde com a esclera. A córnea central é geralmente normal.
Se existirem erros de refração estes são corrigidos.
Podem ser utilizadas lágrimas artificiais.
Na esclerocórnea generalizada, a queratoplastia precoce deve ser considerada para permitir e/ou preservar a visão.
Alterações periféricas da esclera observadas em paciente com esclerocórnea
Imagem: “Sclerocornea” por Mataftsi A et al. Licença: CC BY 3.0
Distrofias da Córnea
Distrofia estromal congénita da córnea
Doença da córnea hereditária, não inflamatória, geralmente não associada a nenhuma outra patologia ocular ou sistémica.
Descrição geral:
Frequentemente envolve a área central da córnea
Áreas discretas de opacificação desenvolvem-se nas camadas superficiais do estroma.
Etiologia:
Causada por mutações no geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics DCN
Autossómica dominante
Classificação:
Distrofia reticular tipo I e II
Distrofia granular tipo I e II
Distrofia macular
Distrofia Reis-Bucklers
Fisiopatologia:
Os sintomas clínicos surgem pela atrofia e degeneração das células epiteliais basais.
Depósitos amilóides e hialinos também contribuem para o desenvolvimento dos sintomas.
Há um aumento no número e na densidade até que a membrana de Bowman sofre erosão e o epitélio descama.
As distrofias da córnea podem apresentar-se sob a forma granular, reticular ou macular.
A superfície da córnea é normal ou ligeiramente irregular; ao longo do estroma de toda a córnea são encontradas pequenas opacidades que fornecem uma aparência turva.
Apresentação clínica e diagnóstico:
Redução da visão devido aos depósitos
Estrabismo é comum.
Exame com lâmpada de fenda: a córnea anterior apresenta opacidades escamosas ou em forma de pena.
Tratamento:
Óculos ou lentes de contacto para correção de erros refrativos
Adesivos ou correção cirúrgica para o estrabismo
Queratoplastia
Distrofia endotelial congénita da córnea (CHED, pela sigla em inglês)
A distrofia endotelial hereditária congénita é uma distrofia corneana caracterizada por turvação difusa bilateral da córnea.
Etiologia e epidemiologia:
Autossómica recessiva
Mutação no cromossoma 20p13
GeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics afetado: SLC4A11
Incidência: 3 em cada 100.000 nascimentos
Descendentes de casamentos consanguíneos apresentam uma incidência comparativamente maior.
Classificação:
CHED
Distrofia corneana polimorfa posterior (PPCD, pela sigla em inglês)
Fisiopatologia:
O geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics SLC4A11 codifica uma proteína transmembranar, que atua como uma bomba entre o endotélio e o estroma da córnea.
A disfunção desta proteína leva à turvação e deposição de material nas camadas da córnea.
Apresentação clínica e diagnóstico:
História de turvação bilateral congénita ou perinatal da córnea
Resultados do exame objetivo:
Diminuição da visão
Ambliopia
Nistagmo
Tratamento:
A cirurgia é a base do tratamento.
As cirurgias realizadas são a queratoplastia penetrante ou a queratoplastia endotelial com “strippping” da membrana de Descemet.
Patologias Adquiridas da Córnea
Queloides da córnea
Queloides da córnea são lesões raras, benignas e branco-acinzentadas da córnea que resultam da proliferação anormal de tecido fibroso. A acumulação de colagénio e de diversas glicoproteínas resulta em hiperplasia do epitélio e rutura da camada de Bowman.
Descrição geral:
Congénito: existem casos de queloides da córnea bilaterais associados à síndrome de Lowe e à síndrome de Rubinstein-Taybi.
Adquirido: pode desenvolver-se após trauma ocular ou infeção
Relatado em idades dos 2 meses aos 72 anos, mas globalmente raro
A maioria dos queloides da córnea ocorre nas primeiras 3 décadas de vida.
Patogénese: O sobrecrescimento do estroma da córnea durante o processo de cicatrização resulta na transformação dos queratócitos em fibroblastos e miofibroblastos, levando à formação do queloide.
Apresentação clínica:
A maioria dos pacientes apresenta uma lesão da córnea de crescimento lento que parece surgir da superfície da córnea.
Aparência branco-acinzentada
Perda de visão progressiva e indolor
Impossibilidade de encerrar as pálpebras no caso de queloides de grandes dimensões
O diagnóstico é realizado através da avaliação da acuidade visual e do exame com lâmpada de fenda.
Tratamento:
Sem necessidade de tratamento, se assintomático
Monitorização apertada:
Tamanho do queloide
Acuidade visual
Ponderar cirurgia quando o queloide provoca diminuição da acuidade visual:
Excisão local
Queratectomia lamelar superficial ou fototerapêutica
Queratoplastia
Os queloides da córnea surgem como opacidades que emergem através da superfície da epicórnea.
Imagem: “Slit-lamp microscopic photographs of corneal keloids” por BMC Ophthalmology/Hyo Kyung Lee et al. Licença: CC BY 4.0, recortado por Lecturio.
Queratopatia em banda
Esta é uma alteração degenerativa caracterizada pela presença de depósitos de sais de cálcio na membrana de Bowman e na lamela anterior do estroma do epitélio corneano.
Descrição geral:
A deposição pode ocorrer por diversos fatores:
Precipitação de lágrimas
Alterações do pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance
Compromisso da função endotelial
EdemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema da córnea
A restante córnea permanece transparente.
Apresenta-se como uma opacidade em forma de banda na zona interpalpebral, com um intervalo transparente entre as extremidades da banda e o limbo.
Causado por inflamação ocular crónica devido a:
Uveíte
Queratouveíte herpética crónica
Phthisis bulbi
Queratoconjuntivite seca
Artrite idiopática juvenil
Queratite por herpes zosterHerpes ZosterVaricella-zoster virus (VZV) is a linear, double-stranded DNA virus in the Herpesviridae family. Shingles (also known as herpes zoster) is more common in adults and occurs due to the reactivation of VZV. Varicella-Zoster Virus/Chickenpox
Pode estar associada a distúrbios sistémicos:
Hiperparatiroidismo
Toxicidade pela vitamina D
Doença renal crónica
Hipofosfatasia
Sarcoidose
Apresentação clínica:
A diminuição da acuidade visual é diretamente proporcional à quantidade de cálcio depositado na córnea.
Sensação de corpo estranho
Irritação ocular com hiperemia conjuntival
Fotofobia
A deposição de cálcio inicia-se na periferia e progride para dentro.
Tratamento:
Desbridamento superficial e queratectomia lamelar
Tratamento da causa subjacente, se conhecida, de forma a diminuir a deposição de cálcio na córnea.
A Lecturio Medical complementa o teu estudo através de métodos de ensino baseados em evidência, vídeos de palestras, perguntas e muito mais – tudo combinado num só lugar e fácil de usar.
User Reviews
Details
×
Obtenha Premium para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características
Obtenha Premium para ver todos os vídeos
Verifique agora o seu e-mail para obter um teste gratuito.
Crie uma conta gratuita para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características - incluindo o Qbank de Lecturio com perguntas actualizadas ao estilo do board-.