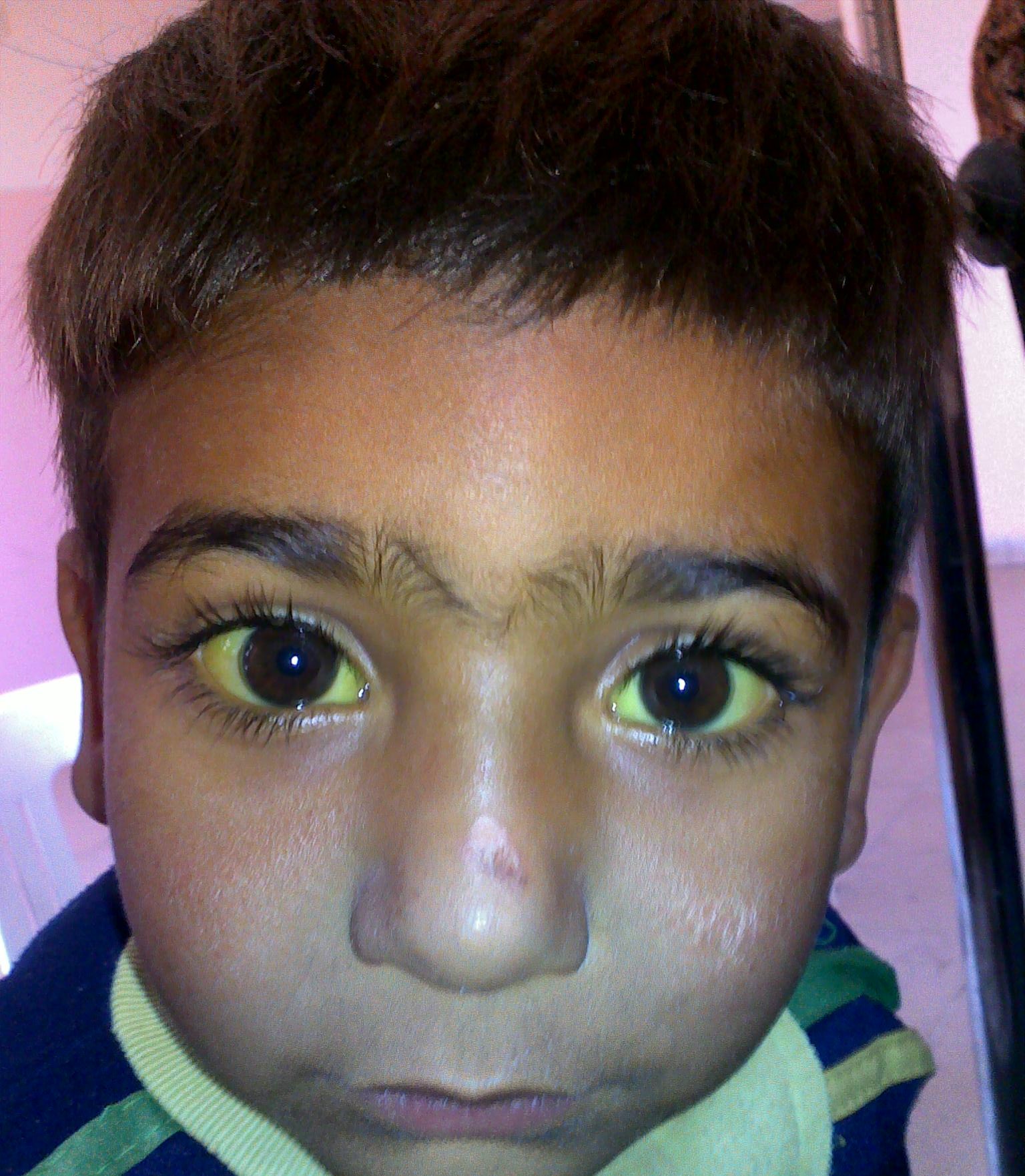



Anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica (AH) é o termo dado a um grande grupo de anemias que são causadas pela destruição prematura/hemólise de glóbulos vermelhos circulantes (GV). A hemólise pode ocorrer dentro dos vasos sanguíneos (hemólise intravascular) ou fora (hemólise extravascular). A destruição extravascular das hemácias é afetada pelos macrófagos do fígado, baço, medula óssea e gânglios linfáticos. Além do local de destruição, a AH pode ser também classificada de acordo com o tipo de defeito do GV, que causa sua destruição. Se o GV tem um defeito intrínseco e geralmente herdado, como uma hemoglobinopatia, um defeito de membrana ou um defeito metabólico, a sua destruição é chamada de hemólise intracorpuscular. Se o GV é normal mas é danificado por uma força externa, como um anticorpo, trauma mecânico ou um agente patogénico, então a sua destruição é classificada como hemólise extracorpuscular, que é quase sempre uma patologia adquirida.
Last updated: Dec 15, 2025
Hemólise intravascular
Hemólise extravascular
A anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica é uma anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types normocítica.
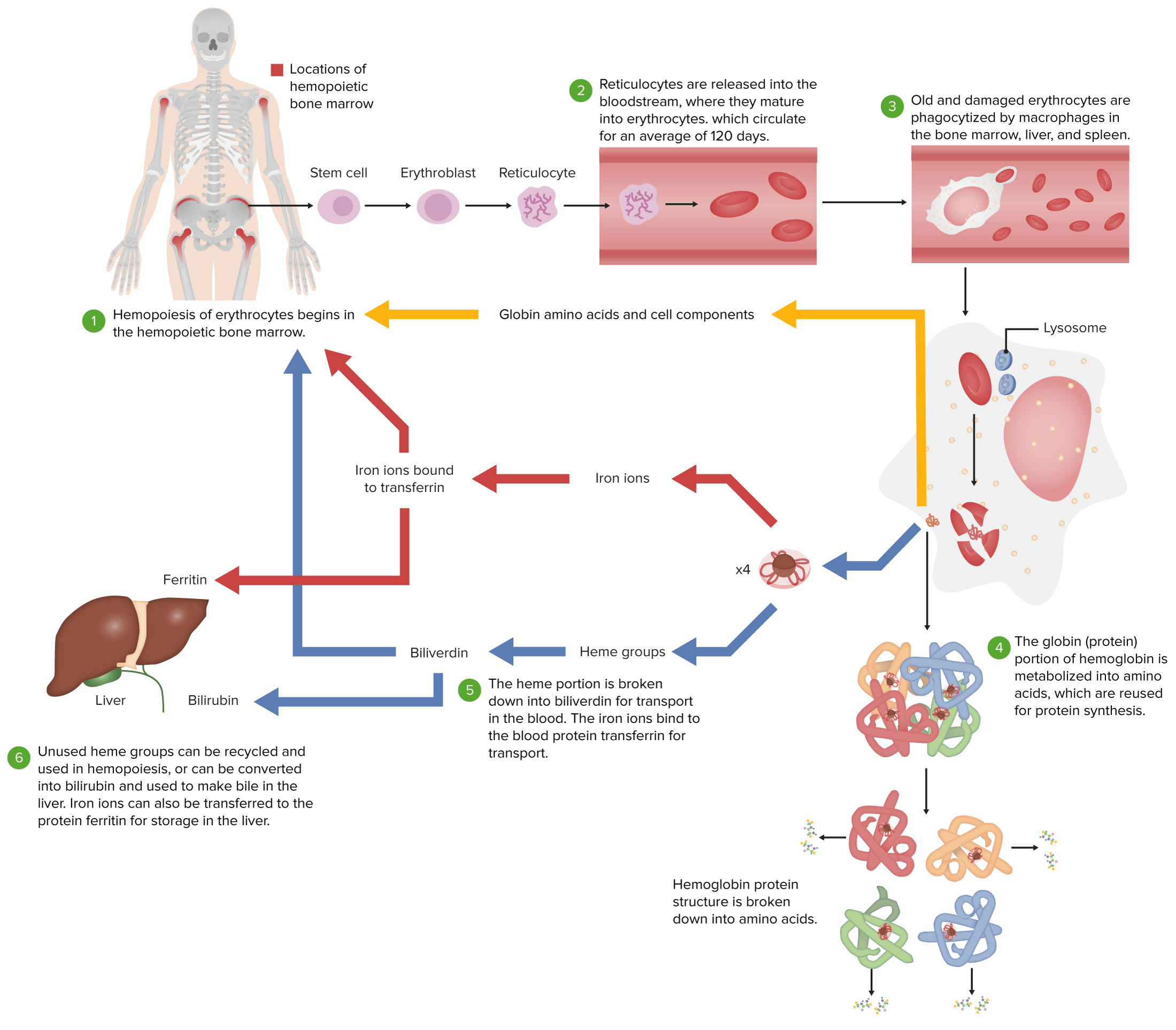
Ciclo de vida normal de um eritrócito
Imagem por Lecturio.Geral
Hemólise intravascular
Hemólise extravascular
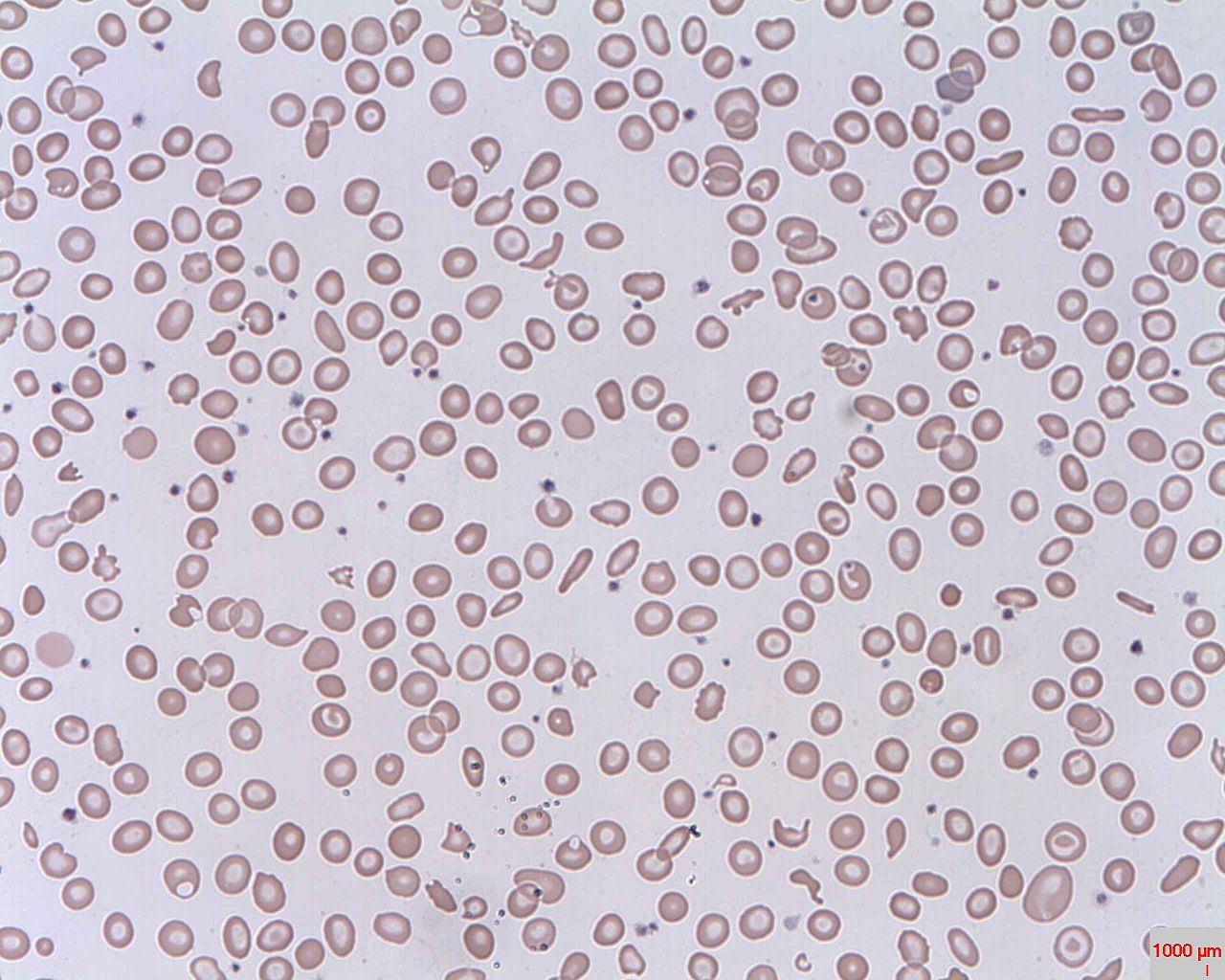
Esquizócitos (GV fragmentados) – hemólise intravascular
Imagem: “Schistocytes” de Prof. Osaro Erhabor. Licença: Domínio Público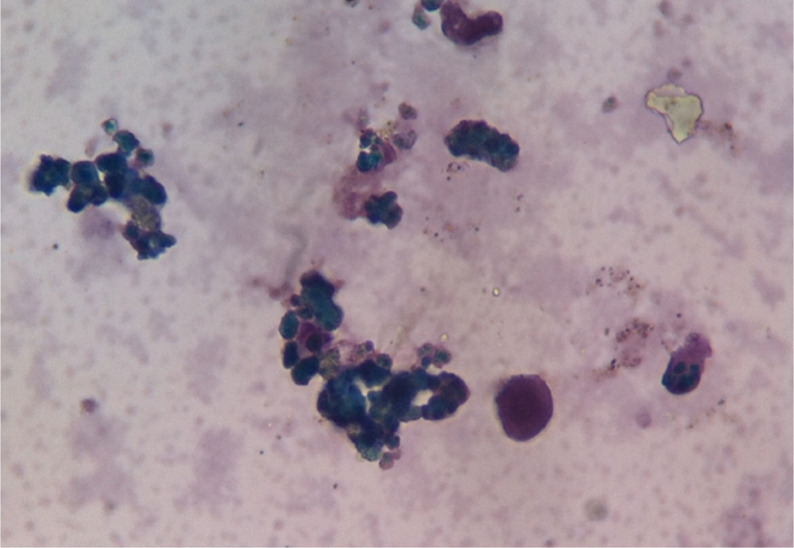
Hemosiderinúria – hemólise intravascular
Imagem: “Hemosiderinuria. Perls’ reaction weakly positive in the urine.” de Salido, Eduardo & Cabañas, Valentín & Berenguer, Mercedes & Macizo, María & Candel, Faustino & Pérez-López, Raúl & Moraleda, Jose. (2014). Achados serológicos numa criança com Hemoglobinúria Paroxística Fria. Relatos de caso em medicina. 2014. Editado por Lecturio.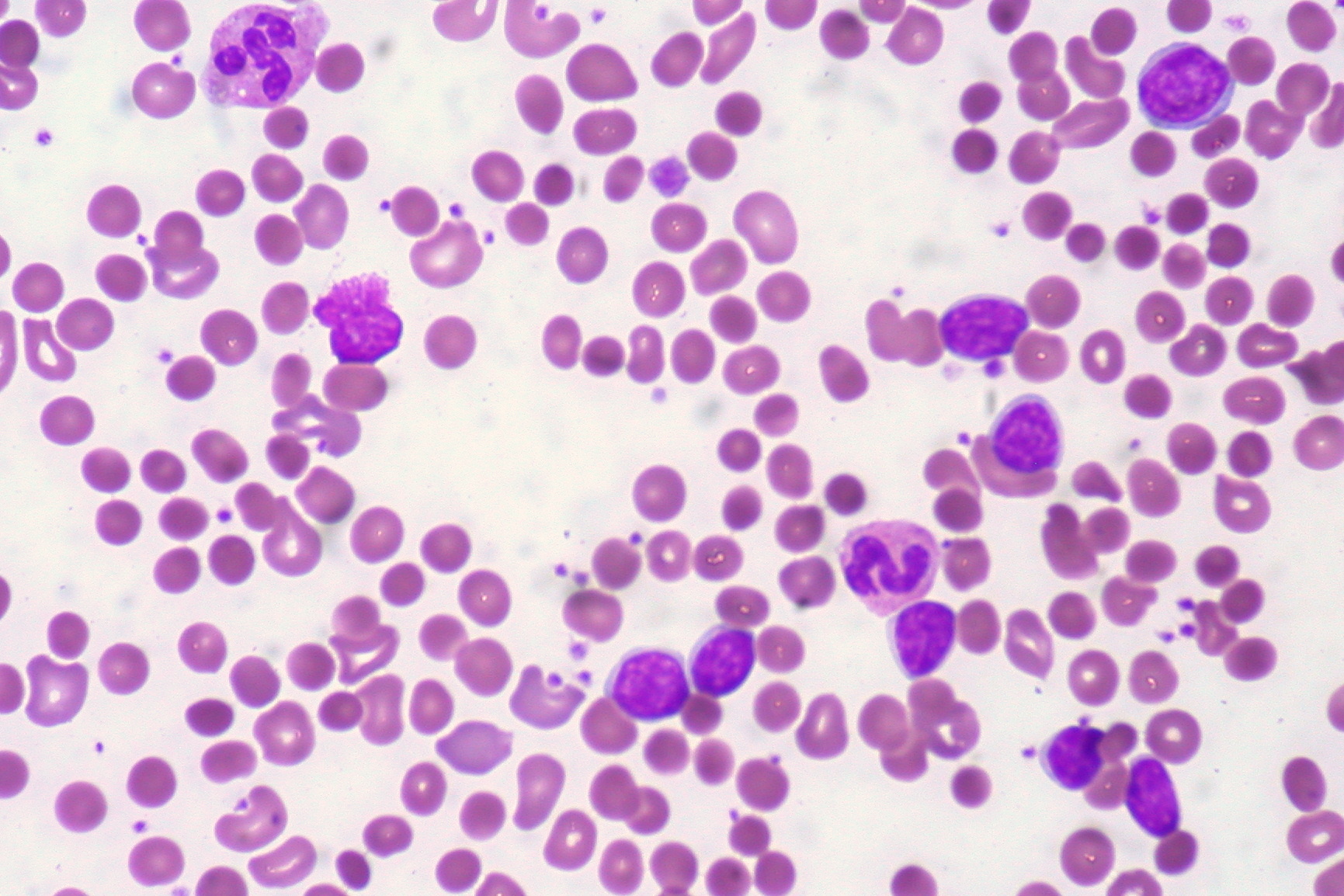
Paciente com LLC apresenta-se com AHAI quente. O esfregaço de sangue mostra esferócitos.
Imagem: “CLL with Autoimmune Hemolytic Anemia” de Ed Uthman from Houston, TX, USA. Licença: CC BY 2.0