


As síndromes mielodisplásicas (SMD) são um grupo de neoplasias clonais com defeitos de maturação caracterizadas por displasia, citopenia e precursores imaturos da medula óssea. As síndromes mielodisplásicas podem ser idiopáticas ou secundárias a várias exposições prejudiciais, como quimioterapia citotóxica, radiação ionizante ou toxinas ambientais. A idade média dos doentes é de 70 anos. A apresentação inclui sintomas de anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (fadiga), neutropenia Neutropenia Neutrophils are an important component of the immune system and play a significant role in the eradication of infections. Low numbers of circulating neutrophils, referred to as neutropenia, predispose the body to recurrent infections or sepsis, though patients can also be asymptomatic. Neutropenia (infeção) ou trombocitopenia (hemorragia). O diagnóstico é baseado na avaliação da medula óssea, que revela citopenia, displasia em pelo menos 1 linhagem e células blásticas em < 20% da celularidade medular. São necessários estudos citogenéticos e moleculares para a classificação, prognóstico e decisões relacionadas com a terapêutica. Há um risco cumulativo aumentado de transformação em LMA, o qual varia dependendo do subtipo de SMD. O tratamento inclui cuidados de suporte, uso de fatores de crescimento hematopoiéticos, terapêutica imunossupressora e transplante alogénico de células hematopoiéticas.
Last updated: Dec 15, 2025
As síndromes mielodisplásicas (SMD) são doenças clonais da medula óssea caracterizadas pela presença de precursores medulares displásicos imaturos e citopenias periféricas. A Organização Mundial de Saúde (OMS) publicou uma atualização em 2022, na qual passou a designar as síndromes mielodisplásicas por “neoplasias mielodisplásicas (SMD)”.
A maioria das SMD são atribuídas a deleções e translocações cromossómicas, ou mutações genéticas.
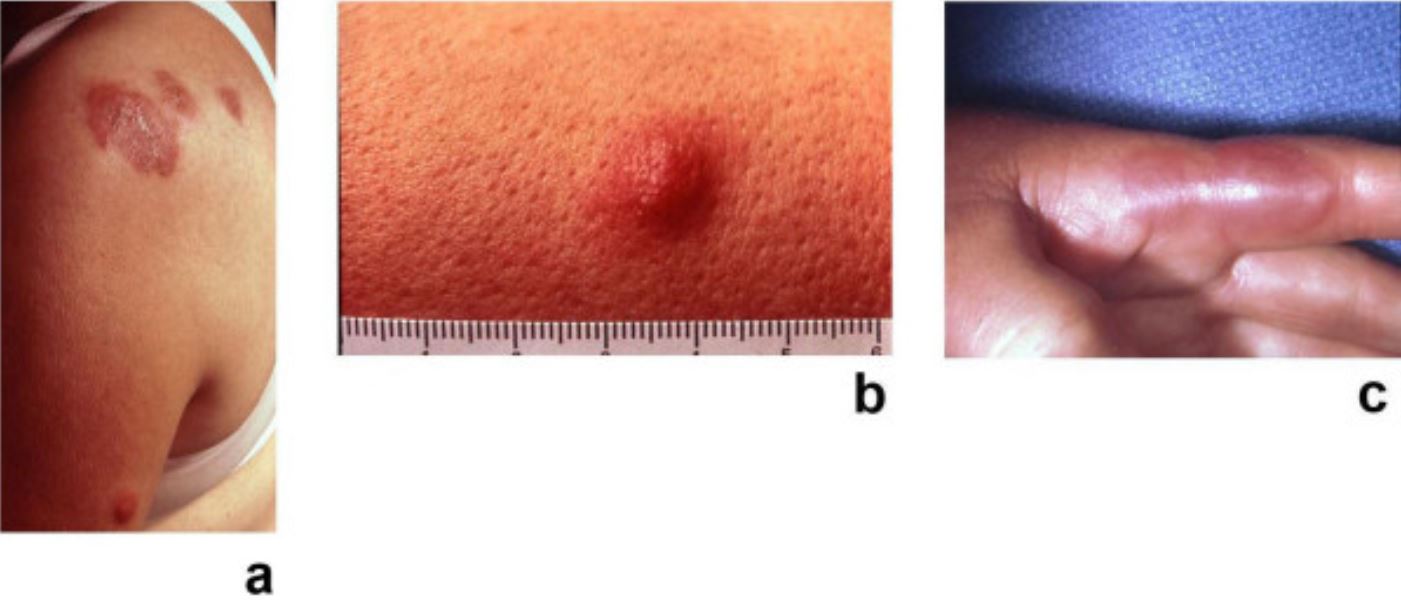
Síndrome de Sweet: placas dolorosas, eritematosas e pseudovasculares da dermatose neutrofílica febril aguda
Imagem: “Sweet’s syndrome–a comprehensive review of an acute febrile neutrophilic dermatosis” por Cohen PR. Licença: CC BY 2.0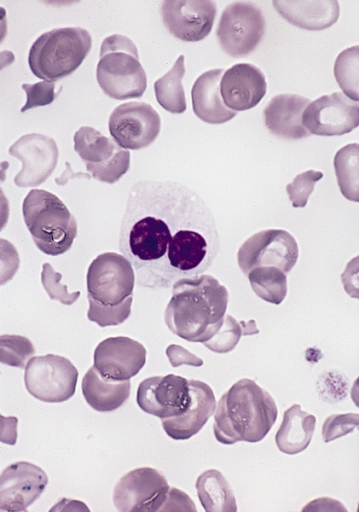
Esfregaço periférico na SMD a demostrar um neutrófilo displásico com núcleo bilobado e alteração da granulação (pseudo-alteração de Pelger-Huët)
Imagem: “Hypogranular neutrophil with a pseudo-Pelger-Huet nucleus in MDS” por The Armed Forces Institute of Pathology (AFIP). Licença: Public Domain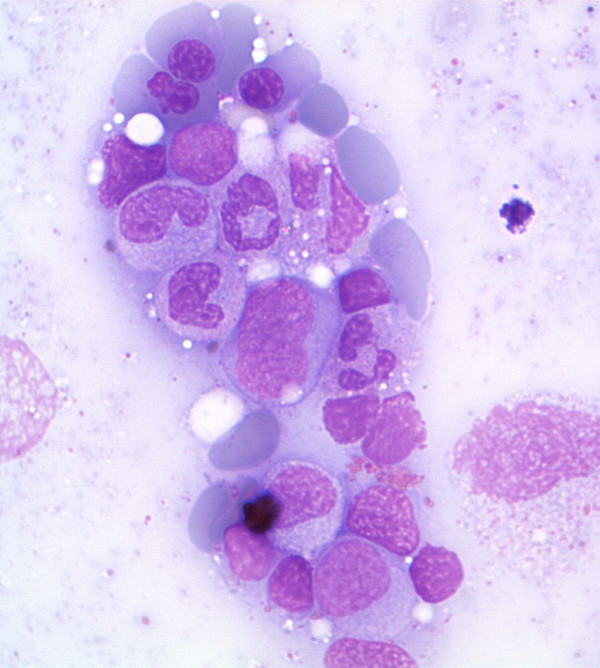
Aspirado de medula óssa na SMD a demostrar células eritroides displásicas e precursores mieloides
Imagem: “Antiretroviral activity of 5-azacytidine during treatment of a HTLV-1 positive myelodysplastic syndrome with autoimmune manifestations” por Diamantopoulos, P.T. et al. Licença: CC BY 2.0Após a confirmação da SMD, os doentes são categorizados com base na 5.ª edição da Classificação de Tumores Hematolinfóides da OMS, publicada em 2022.
A classificação é baseada em:
Tipos:
