


A síndrome nefrítica configura uma ampla categoria de doenças glomerulares caracterizada por hematúria glomerular, perda variável da função renal e hipertensão. Essas características contrastam com as da síndrome nefrótica, que inclui doenças glomerulares caracterizadas por proteinúria grave, embora às vezes exista sobreposição de > 1 doença glomerular no mesmo indivíduo. As apresentações clínicas da síndrome nefrítica são muito variáveis, desde assintomáticas com alterações urinárias até doença crítica com risco de vida. O diagnóstico é sugerido pela presença de hematúria, proteinúria leve a moderada e algumas serologias (e.g., ANCA ANCA Group of systemic vasculitis with a strong association with anca. The disorders are characterized by necrotizing inflammation of small and medium size vessels, with little or no immune-complex deposits in vessel walls. Rapidly Progressive Glomerulonephritis); a biópsia renal é necessária na maioria dos casos. O tratamento varia tão amplamente quanto as apresentações clínicas, desde uma atitude vigilante em casos leves até à imunossupressão e plasmaférese na doença agressiva.
Last updated: Dec 15, 2025
A síndrome nefrítica é definida por alguns ou todos os achados seguintes:

Microscopia da urina na síndrome nefrítica:
Esquerda: eritrócito normal com aparência circular
Direita (3 imagens): eritrócitos dismórficos com bolhas nas membranas e células “Mickey Mouse”
Os 2 principais mecanismos de lesão glomerular nas síndromes nefríticas são a mediação por imunocomplexos e a desregulação do complemento. As formas hereditárias de GN (e.g., síndrome de Alport) têm fisiopatologias diferentes.
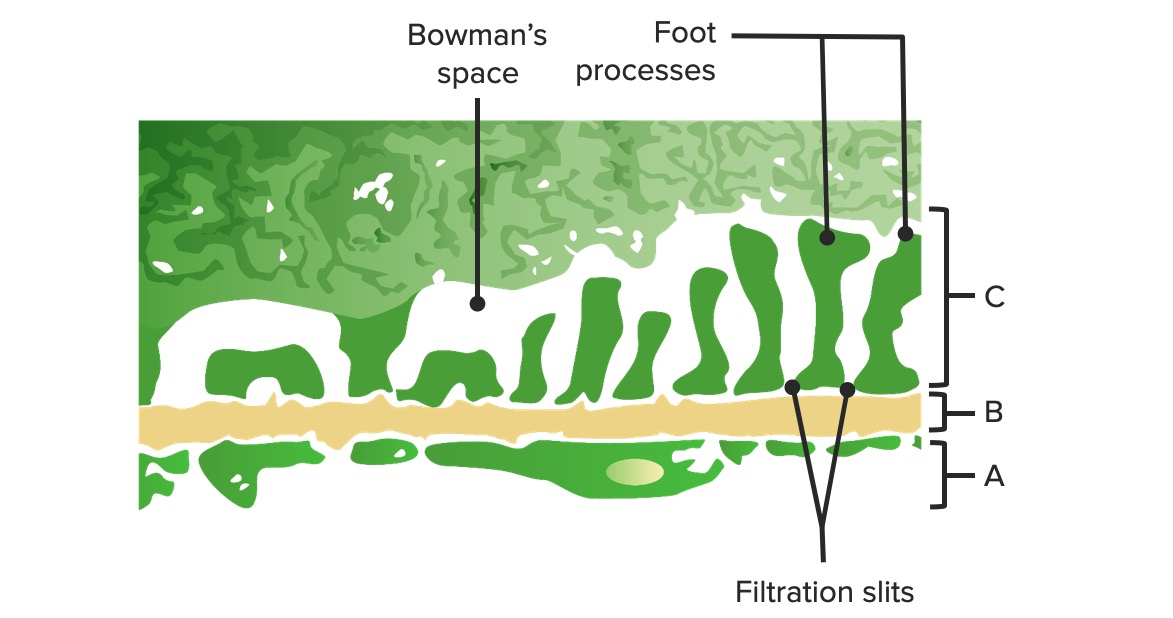
Diagrama da barreira glomerular:
A: Endotélio fenestrado dos capilares glomerulares
B: Membrana basal
C: Camada epitelial a mostrar os processos podocitários e as proteínas estruturais criando o diafragma em fenda

Vias primárias do sistema do complemento
MBL, pela sigla em inglês: linfocitose monoclonal de células B
As causas primárias e secundárias de GN variam de acordo com a etiologia, mas têm em comum a hematúria e graus variáveis de proteinúria. Os achados da biópsia diferem e são importantes saber.
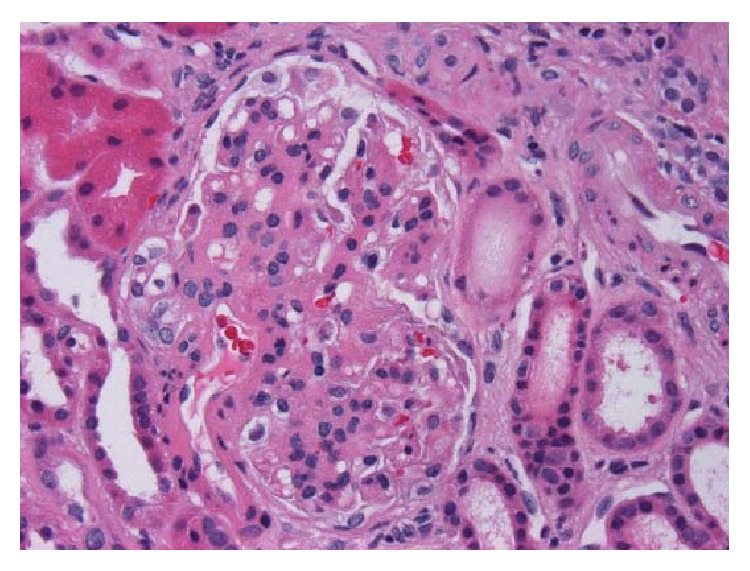
Biópsia renal com nefropatia por IgA:
H&E, x400. Glomérulo com mesângio espessado e hipercelularidade mesangial segmentar
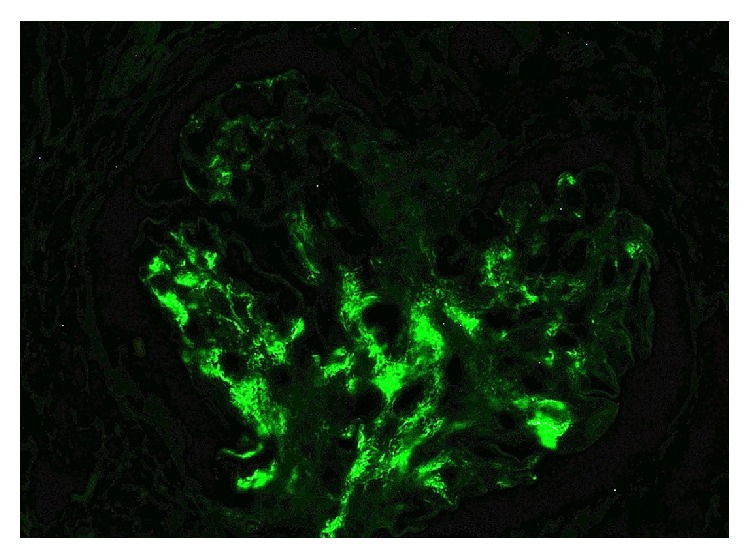
Nefropatia de IgA:
Coloração de imunofluorescência a demonstrar positividade para IgA
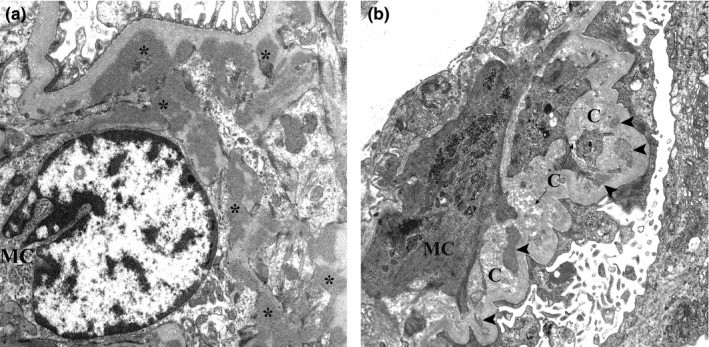
Micrografia eletrónica de biópsia renal de nefropatia por IgA:
(a): Vários depósitos mesangiais eletrodensos (asteriscos) são mostrados. Esses depósitos não possuem o halo lucente do eletrão.
(b): São evidentes depósitos mesangiais com graus variados de reabsorção (pontas de seta) próximos às fibras de colagénio.
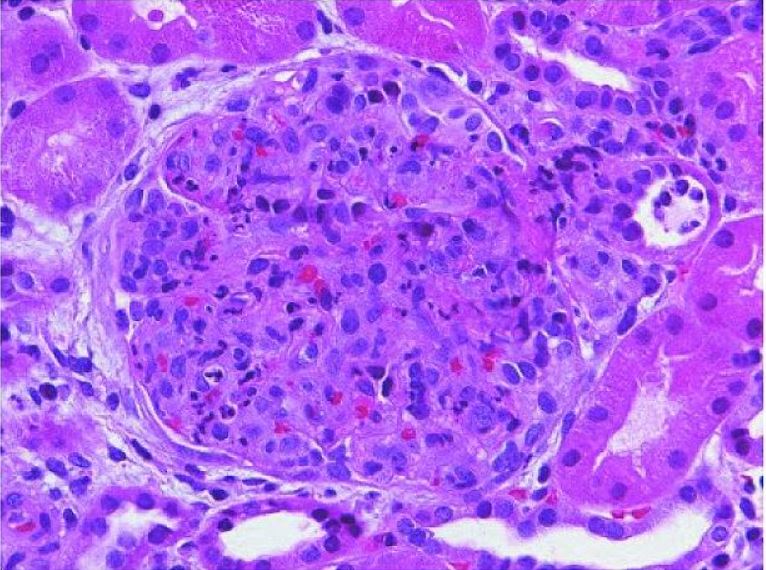
Glomerulonefrite pós-estreptocócica aguda:
Glomérulo aumentado com encerramento global do lúmen capilar causado por proliferação de células predominantemente mesangiais endógenas e infiltração de monócitos e leucócitos polimorfonucleares (coloração H&E).
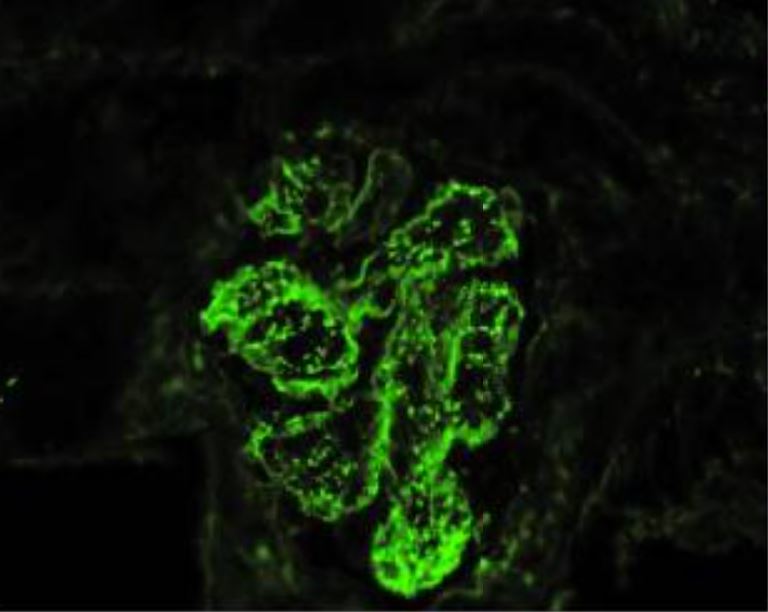
Glomerulonefrite aguda pós-estreptocócica IgA com febre reumática concomitante (microscopia de imunofluorescência):
mostra depósitos imunes na parede dos capilares glomerulares e mesângio granular com um padrão de “céu estrelado” com dominância de IgA
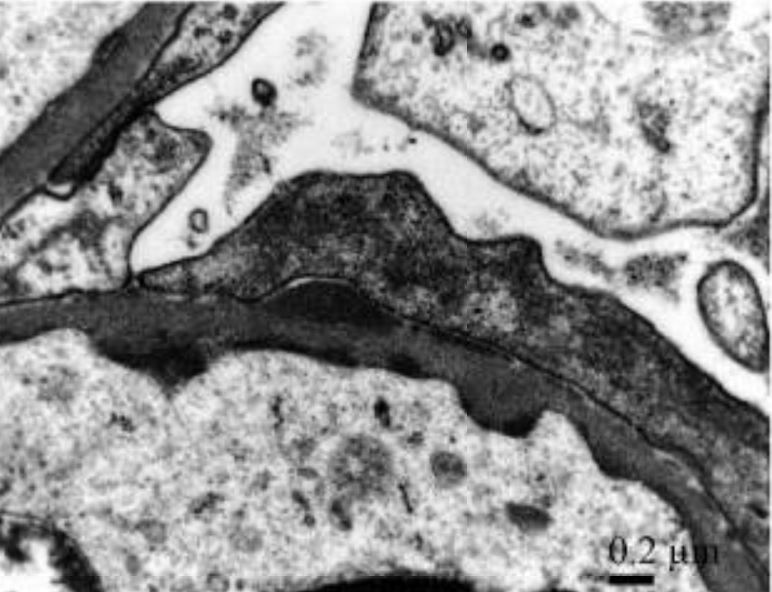
Glomerulonefrite aguda pós-estreptocócica IgA com febre reumática concomitante:
Depósitos subendoteliais e subepiteliais eletrodensos discretos detetados usando alta ampliação que mostram depósitos imunes granulares mesangiais e glomerulares na parede capilar com um padrão de “céu estrelado” com dominância de IgA
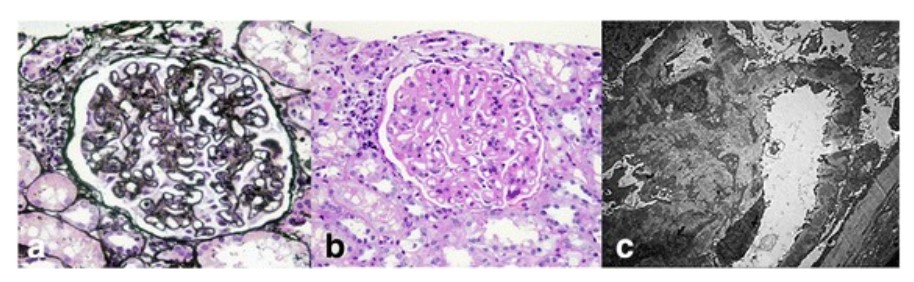
Achados histopatológicos renais no lúpus eritematoso sistémico:
Micrografias de luz de um glomérulo mostram espessamento difuso da membrana basal glomerular (coloração ácida periódico-metenamina de prata; × 400) (a) com hipercelularidade mesangial e expansão da matriz mesangial (coloração de ácido periódico-Schiff; × 400) (b). Análises de microscopia eletrónica mostram numerosos depósitos eletrodensos subepiteliais e mesangiais, bem como depósitos dentro da membrana basal glomerular com extensa fusão dos processos podocitários (×5000) (c).
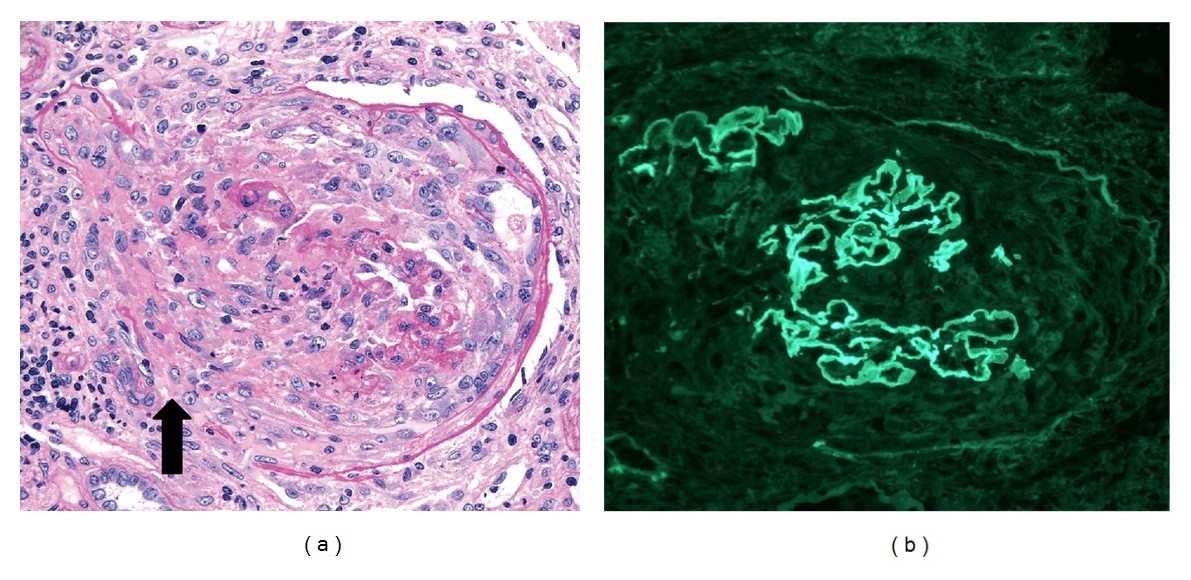
Glomérulos de uma biópsia renal de glomerulonefrite crescêntica, com antimembrana basal glomerular e anticorpos p-ANCA:
(a): A microscopia de luz mostra crescentes celulares destrutivos com descontinuidades na cápsula de Bowman (seta; coloração de ácido periódico-Schiff, × 250). O interstício adjacente apresenta um infiltrado leucocitário mononuclear.
(b) Coloração de imunofluorescência (×250) mostra uma coloração linear forte (3–4+) de IgG ao longo das paredes capilares. As paredes capilares sofrem disrupção de forma focal devido à formação do crescente.
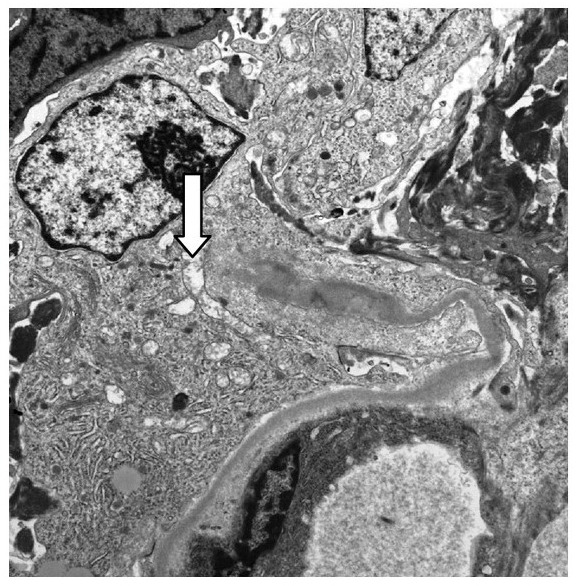
Micrografia eletrónica × 10.000 a mostrar uma parede capilar glomerular quebrada (seta) associada a um crescente celular na doença de Goodpasture
Imagem: ““Glomeruli from the renal biopsy” por Tariq Javed and Parag Vohra. Licença: CC BY 3.0, recortado por Lecturio.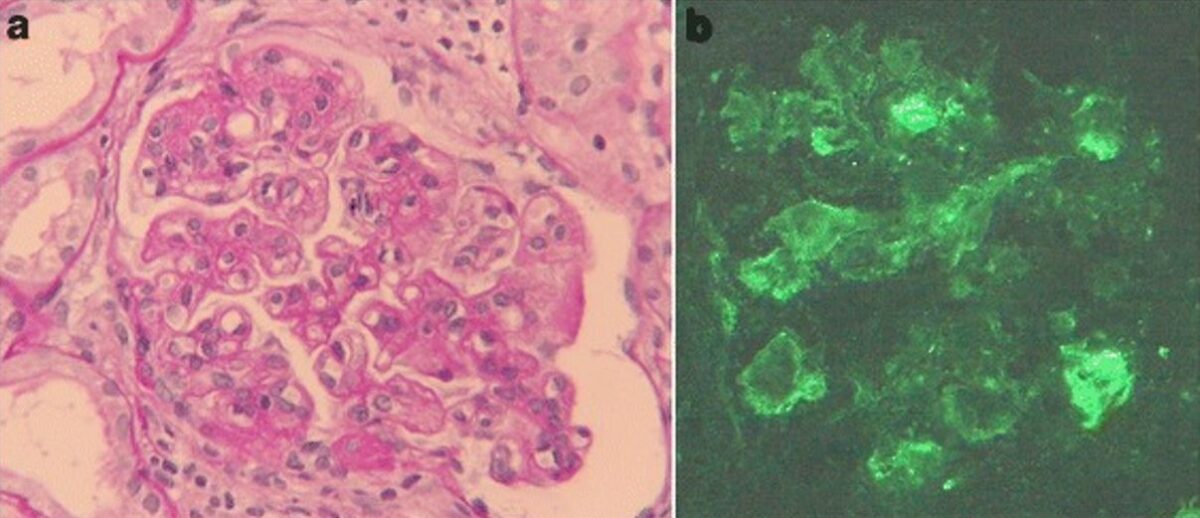
Glomerulonefrite membranoproliferativa associada à hepatite C
a) Achados de microscopia de luz na biópsia renal: A coloração com ácido periódico de Schiff revela hipercelularidade mesangial, acentuação lobular e contorno duplo da membrana basal (ampliação original, 400×).
b) A coloração de imunofluorescência de IgM é positiva ao longo da ansa capilar (ampliação original, ×400).
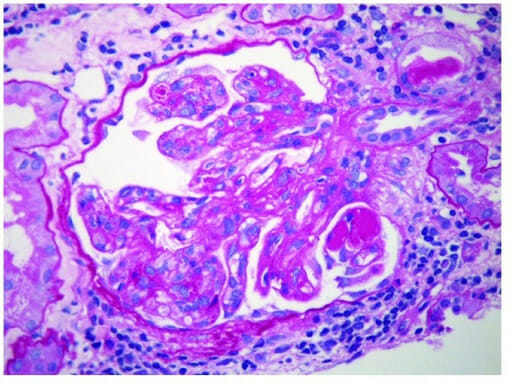
Glomerulonefrite membranoproliferativa:
Micrografia eletrónica de GNMP a mostrar depósitos imunes subendoteliais
