


A síndrome de Zellweger (SZW), também denominada de síndrome cerebrohepatorrenal, é um distúrbio congénito raro da biossíntese dos peroxissomas. É considerada um erro inato do metabolismo e é a forma mais MAIS Androgen Insensitivity Syndrome grave de um espectro de condições denominadas de distúrbios do espectro de Zellweger (DZS). Caracteriza-se pela redução ou ausência de peroxissomas funcionais. Os sintomas estão presentes desde o nascimento e incluem hipotonia, dificuldades na alimentação, convulsões e determinadas características físicas típicas, nomeadamente características faciais e malformações esqueléticas. Não há cura para SZW. A taxa de sobrevida média é inferior a 1 ano.
Last updated: Dec 15, 2025
As manifestações clínicas da SZW estão presentes ao nascimento.
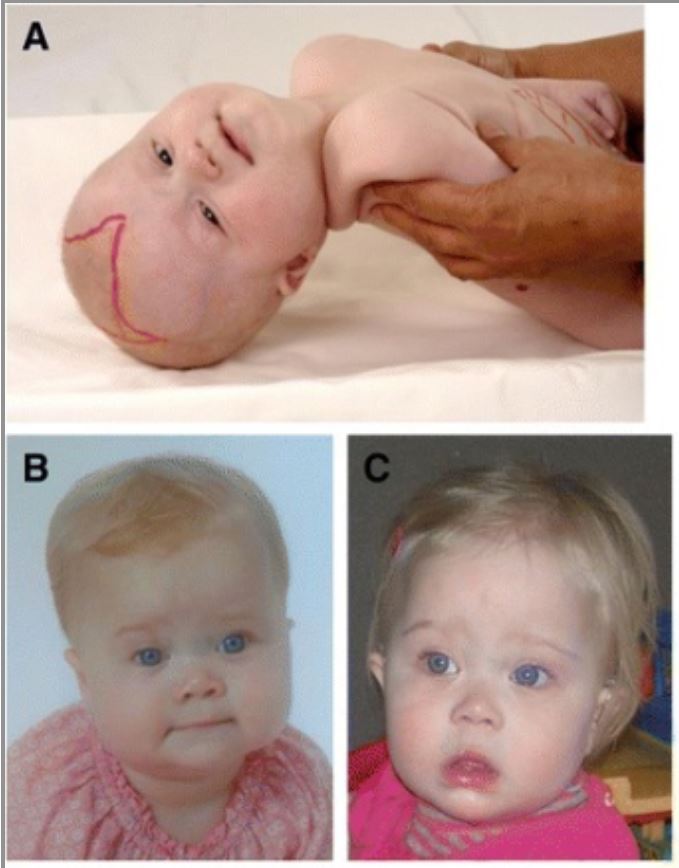
Características dismórficas craniofaciais em doentes com distúrbio do espectro de Zellweger (DZS)
Uma menina de 6 meses (A) com dismorfia craniofacial típica, pregas epicânticas, testa alta, ponte nasal larga, cristas supraorbitais hipoplásicas e fontanela anterior grande (desenhada). As imagens (B) e (C) mostram uma menina com DZS com 9 meses de idade (B) e 14 meses (C). O dismorfismo facial nesta menina é menos pronunciado.
Não há cura nem tratamento eficaz para a SZW. O tratamento é de suporte, na melhor das hipóteses.
A morte geralmente ocorre dentro de 1 ano após o nascimento (geralmente < 6 meses), geralmente devido a insuficiência respiratória.
A síndrome de Zellweger faz parte de um continuum clínico de distúrbios da biogénese do peroxissoma conhecidos como distúrbios do espectro de Zellweger (DZS). Todas as condições a seguir têm, também, hereditariedade autossómica recessiva e são diagnosticadas e abordadas de forma semelhante à ZWS ZWS Zellweger syndrome (ZWS), also called cerebrohepatorenal syndrome, is a rare congenital peroxisome biosynthesis disorder and is considered an inborn error of metabolism. Zellweger syndrome is the most severe form of a spectrum of conditions called Zellweger spectrum disorder (ZSD), and is characterized by the reduction or absence of functional peroxisomes. Zellweger Syndrome:
O diagnóstico diferencial da SZW é vasto. Características clínicas, testes Testes Gonadal Hormones bioquímicos, imagens e, finalmente, testes Testes Gonadal Hormones genéticos ajudam a diferenciar e fornecer um diagnóstico. Os seguintes são distúrbios que devem ser considerados em qualquer recém-nascido que apresente hipotonia profunda:
