El síndrome de Zellweger, también llamado síndrome cerebrohepatorrenal, es un raro trastorno congénito de la biosíntesis de los LOS Neisseria peroxisomas y se considera un error Error Refers to any act of commission (doing something wrong) or omission (failing to do something right) that exposes patients to potentially hazardous situations. Disclosure of Information innato del metabolismo. El síndrome de Zellweger es la forma más grave de un espectro de afecciones denominado trastorno del espectro de Zellweger, y se caracteriza por la reducción o ausencia de peroxisomas funcionales. Los LOS Neisseria síntomas están presentes desde el momento del nacimiento e incluyen hipotonía, mala alimentación, convulsiones y ciertos rasgos físicos distintivos, especialmente características faciales y malformaciones esqueléticas. No hay cura para el síndrome de Zellweger. La tasa media de supervivencia es inferior a un año.
Last updated: Dec 15, 2025
Las manifestaciones clínicas del síndrome de Zellweger están presentes al AL Amyloidosis nacer.
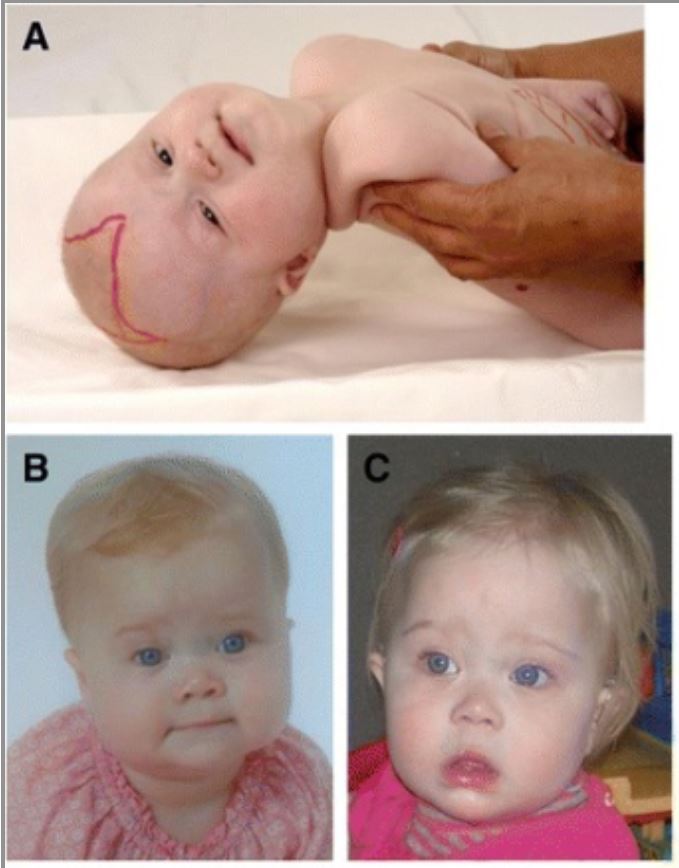
Rasgos dismórficos craneofaciales en pacientes con trastorno del espectro de Zellweger (ZSD)
Una niña de 6 meses (A) con dismorfia craneofacial típica, pliegues epicánticos, frente alta, puente nasal ancho, rebordes supraorbitales hipoplásicos y fontanela anterior grande (dibujada). Imágenes (B) y (C) muestran a una niña con ZSD a la edad de 9 meses (B) y 14 meses (C). El dismorfismo facial en esta niña es menos pronunciado.
No hay cura ni tratamiento eficaz para el síndrome de Zellweger. El tratamiento es de soporte en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el mejor de los LOS Neisseria casos.
La muerte suele producirse en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el plazo de 1 año tras el nacimiento (normalmente < 6 meses), normalmente debido a una insuficiencia respiratoria.
El síndrome de Zellweger forma parte de un grupo clínico de trastornos de la biogénesis del peroxisoma conocido como trastorno del espectro de Zellweger. Todas las siguientes enfermedades también tienen una herencia autosómica recesiva, y se diagnostican y tratan de forma similar al AL Amyloidosis síndrome de Zellweger:
El diagnóstico diferencial del síndrome de Zellweger es muy amplio. Las características clínicas, pruebas bioquímicas, imagenología y, en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum última instancia, las pruebas genéticas ayudan a diferenciar y proporcionar un diagnóstico. Los LOS Neisseria siguientes son trastornos que deben considerarse en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum cualquier neonato que presente una hipotonía profunda: