


A síndrome de Williams (SW), também conhecida como síndrome de Williams-Beuren (SWB), é uma doença genética causada por uma microdeleção no cromossoma 7. Os indivíduos afetados têm uma fácies élfica característica e baixa estatura. São comuns problemas cognitivos, do desenvolvimento e comportamentais. Além disso, também podem estar presentes anomalias cardiovasculares, do tecido conjuntivo, endócrinas e renais. O teste genético confirma o diagnóstico. O tratamento é baseado nas manifestações clínicas. O envolvimento cardiovascular precisa de seguimento apertado, pois é a principal causa de mortalidade.
Last updated: Jan 24, 2025
A incidência da síndrome de Williams (SW) é de 1 em 7.500–20.000 nados vivos.
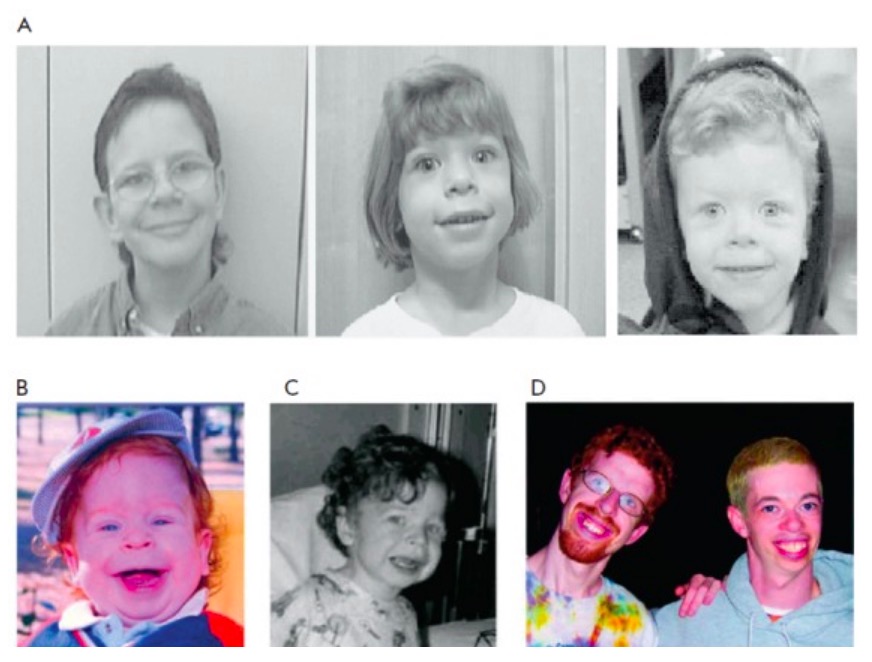
Características faciais da SW
A: aparência facial distinta de indivíduos com SW
B e C: criança pequena com SW aos 15 meses e 3 anos (observar as características faciais subtis, incluindo boca larga, bochechas cheias, filtro longo, nariz pequeno e queixo delicado)
O mesmo doente é mostrado nas Figs. B, C e D (esquerda; aos 21 anos); outro indivíduo com SW aos 28 anos é apresentado na Fig. D à direita.
A cardiopatia congénita está presente em 80%–90% dos casos de SW e é uma das principais causas de morbilidade e mortalidade. O risco de morte súbita cardíaca é 25-100 vezes maior nestes doentes.
O tratamento é baseado nas manifestações clínicas presentes e visa melhorar a qualidade de vida e tratar as condições associadas.
