


O síndrome de Prader-Willi (SPW) e o síndrome de Angelman (SA) são ambos doenças genéticas autossómicas raras do neurodesenvolvimento, ligadas a uma região específica do cromossoma 15, atribuídas ao "imprinting" genómico. Isto significa que o fenótipo depende do sexo dos pais PAIS Androgen Insensitivity Syndrome que doam os genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure. Um cromossoma 15 derivado do pai com esta deleção resulta na síndrome de deleção paterna 15q11-13, ou SPW, enquanto um cromossoma 15 derivado da mãe com uma deleção semelhante está associado ao SA. O diagnóstico é feito através de testes Testes Gonadal Hormones genéticos. O tratamento é principalmente de apoio e está focado na intervenção precoce para o desenvolvimento, anomalias neurológicas e físicas.
Last updated: Dec 15, 2025
Osíndrome de Prader-Willi (SPW) é uma doença associada à perda do cromossoma paterno da região 15q11-13 sendo caracterizado por défice intelectual, baixa estatura, subdesenvolvimento dos órgãos sexuais e obesidade.
O síndrome de Angelman (SA) é uma doença associada à perda do cromossoma materno na região 15q11-13, sendo caracterizado por um grave atraso no desenvolvimento neurológico.
Tanto o SPW como o SA estão associados à impressão genómica, definida como a expressão diferencial de certos genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure baseados na sua transmissão de um progenitor específico.
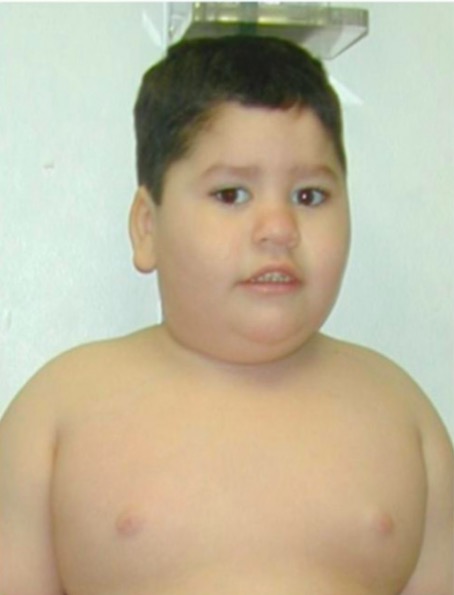
Síndrome de Prader-Willi:
Características faciais típicas e obesidade do tronco
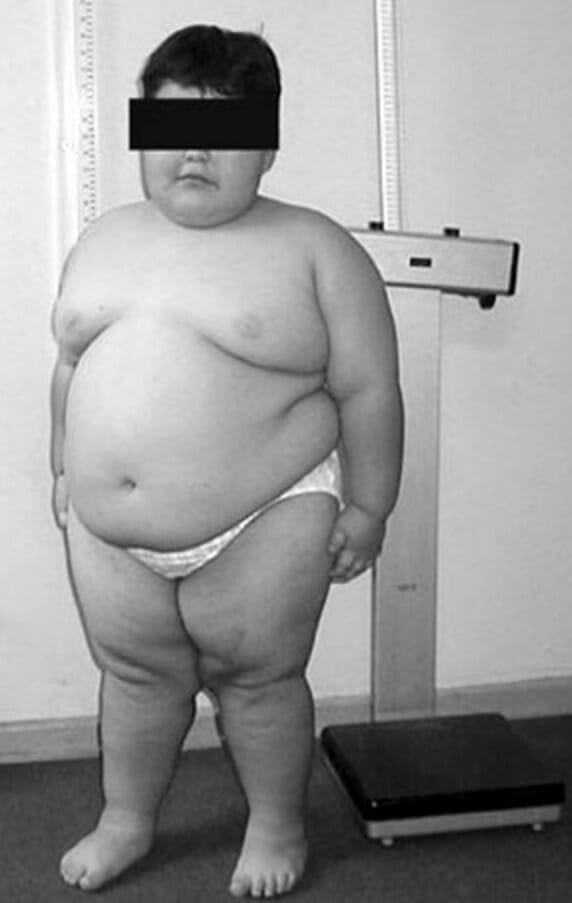
Uma criança com 8 anos com síndrome de Prader-Willi exibindo a obesidade truncal característica
Imagem: “PWS8” por Fanny Cortés M1 et al. Licença: CC BY 4.0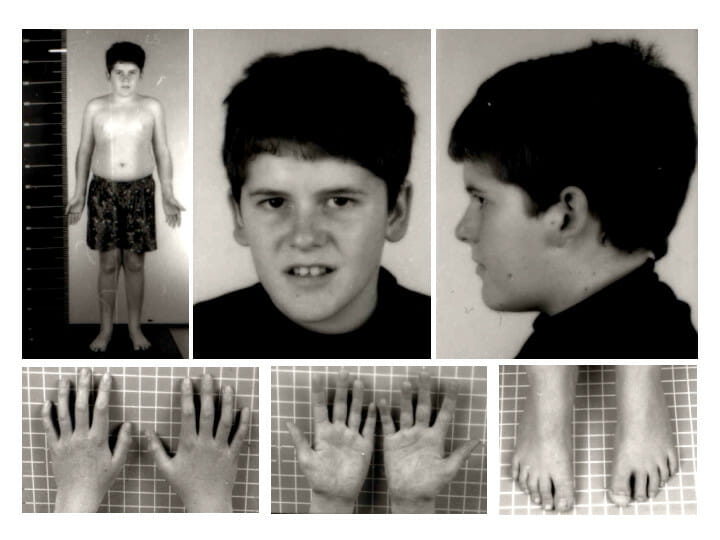
Fenótipo de síndrome de Prader-Willi aos 15 anos de idade: Note a ausência das características faciais típicas da síndrome de Prader-Willi e a presença de obesidade truncal ligeira.
Imagem: “Pws” por Schüle B et al. Licença: CC BY 2.0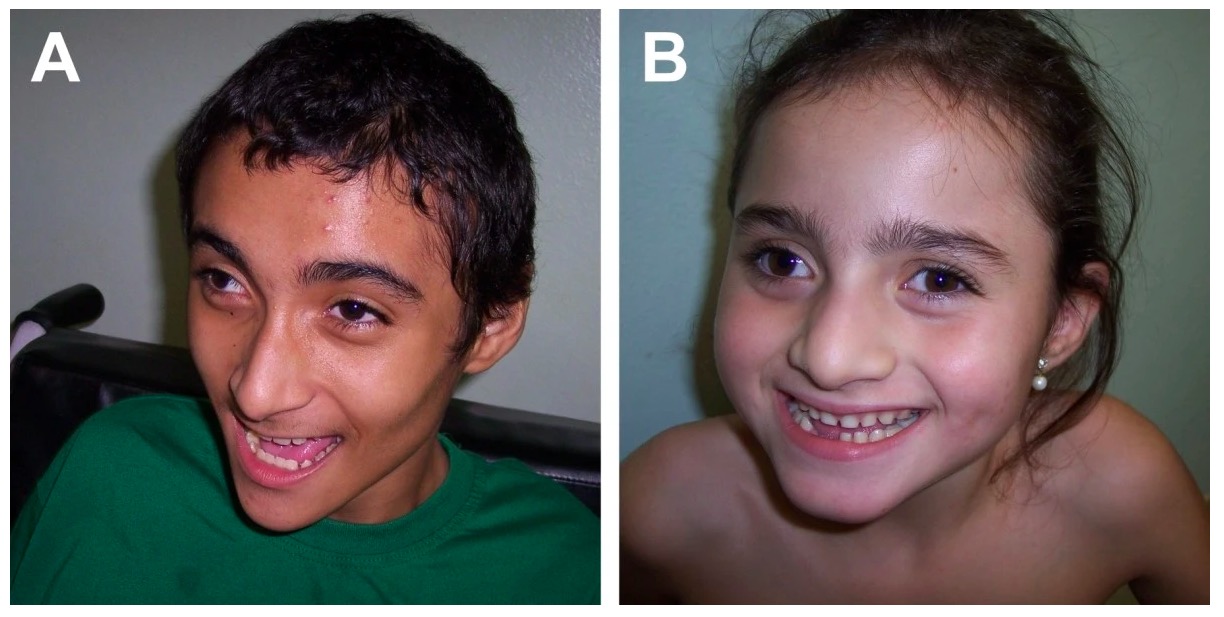
Síndrome de Angelman: características craniofaciais típicas, comportamento feliz
Imagem: “Angelman syndrome” por Department of Genetics, Medical School of Ribeirao Preto, University of Sao Paulo, Sao Paulo, Brazil. Licença: CC BY 2.0