El síndrome de Prader-Willi y el síndrome de Angelman son dos raros trastornos genéticos autosómicos del neurodesarrollo que se relacionan con una región específica del cromosoma 15 debido a la impronta genómica. Esto significa que el fenotipo depende del sexo del progenitor que dona los LOS Neisseria genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure. Un cromosoma 15 de origen paterno con esta deleción da lugar al AL Amyloidosis síndrome de deleción paterna 15q11-13, o síndrome de Prader-Willi, mientras que un cromosoma 15 de origen materno con una deleción similar se asocia con el síndrome de Angelman. El diagnóstico se establece con pruebas genéticas. El tratamiento es principalmente de soporte y se centra en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la intervención temprana de las anomalías del desarrollo, neurológicas y físicas.
Last updated: Dec 15, 2025
El síndrome de Prader-Willi es una enfermedad asociada a la pérdida de la región cromosómica paterna 15q11-13 y se caracteriza por discapacidad intelectual, baja estatura, subdesarrollo de los LOS Neisseria órganos sexuales y obesidad.
El síndrome de Angelman es un trastorno asociado a la pérdida de la región materna del cromosoma 15q11-13 y se caracteriza por un grave retraso del neurodesarrollo.
Tanto el síndrome de Prader-Willi como el síndrome de Angelman están asociados a la impronta genómica, que es la expresión diferencial de ciertos genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure basada en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la herencia de un progenitor específico.
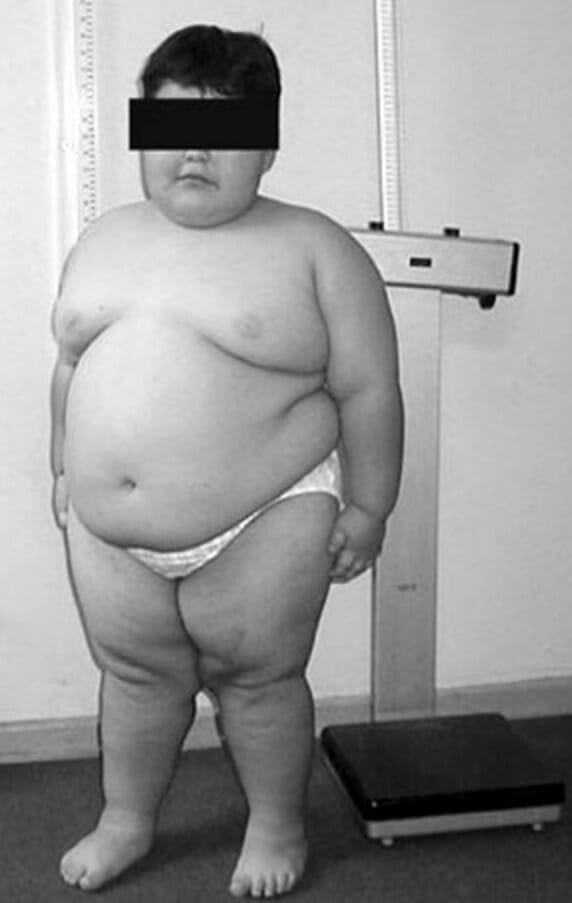
Síndrome de Prader-Willi:
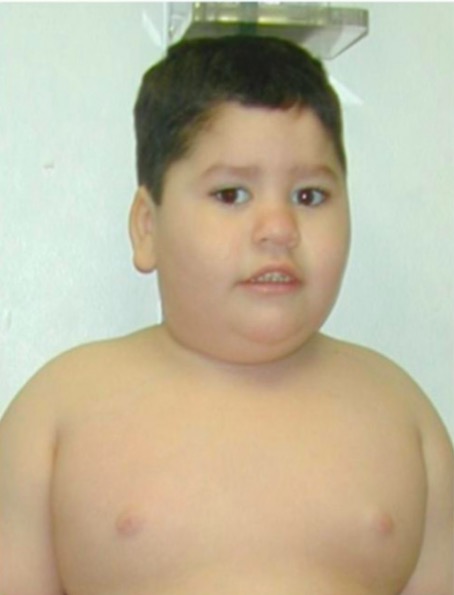
Rasgos faciales típicos y obesidad troncal
Un niño de 8 años con síndrome de Prader-Willi que presenta una obesidad troncal característica
Imagen: “PWS8” por Fanny Cortés M1 et al. Licencia: CC BY 4.0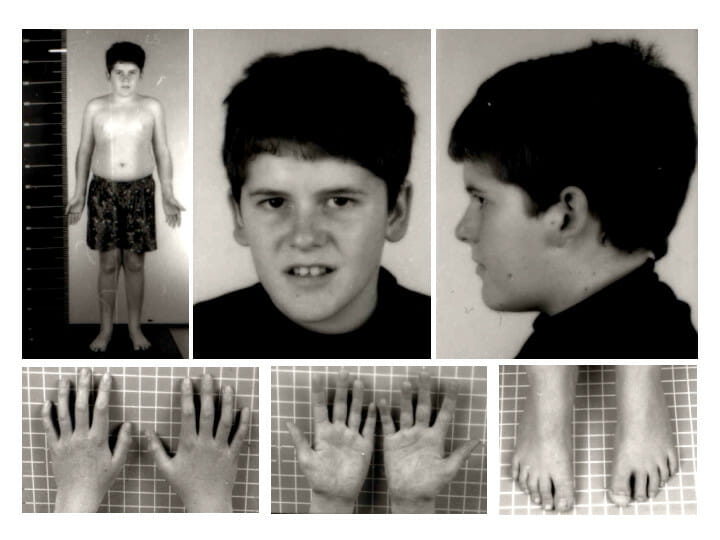
Fenotipo del síndrome de Prader-Willi a los 15 años de edad: Nótese la ausencia de rasgos faciales típicos del síndrome de Prader-Willi y la presencia de una leve obesidad troncal.
Imagen: “Pws” por Schüle B et al. Licencia: CC BY 2.0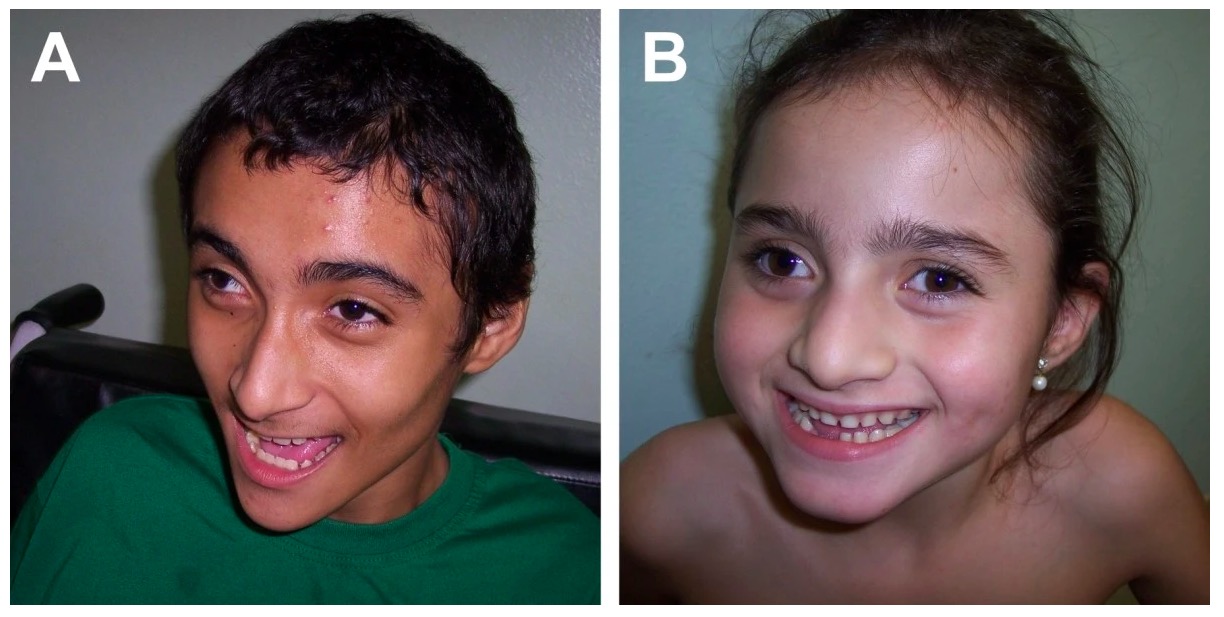
Síndrome de Angelman: rasgos craneofaciales típicos, comportamiento alegre
Imagen: “Angelman syndrome” por Department of Genetics, Medical School of Ribeirao Preto, University of Sao Paulo, Sao Paulo, Brazil. Licencia: CC BY 2.0