


A trissomia 13, ou síndrome de Patau (SP), é um síndrome genético causado pela presença de três cópias do 13.º cromossoma. Sendo a 3.ª trissomia mais MAIS Androgen Insensitivity Syndrome comum, a síndrome de Patau tem uma incidência de 1 em cada 10.000 nados-vivos e é mais MAIS Androgen Insensitivity Syndrome comum nas mulheres. A maioria dos casos de síndrome de Patau são diagnosticados no período pré-natal através de rastreio materno e ecografia. Mais MAIS Androgen Insensitivity Syndrome da metade das gravidezes resulta em abortos espontâneos. Se a gravidez chegar a termo, recomenda-se que um centro especializado trate do parto e dos cuidados neonatais. No recém-nascido, os achados incluem malformações craniofaciais e cardíacas, défice intelectual grave e grande redução da expectativa de vida. A maioria dos bebés não sobrevive para além dos 3 meses. Sem tratamento disponível e com uma expectativa de sobrevivência nula, a família recebe apoio e recursos para lidar com o curso natural da doença.
Last updated: Dec 15, 2025
A trissomia 13 ou síndrome de Patau é definida pela presença de 3 cópias do cromossoma 13.
“Aos 13 anos entras na Puberdade”: P como no síndrome de Patau ou 13 para a trissomia do cromosoma 13
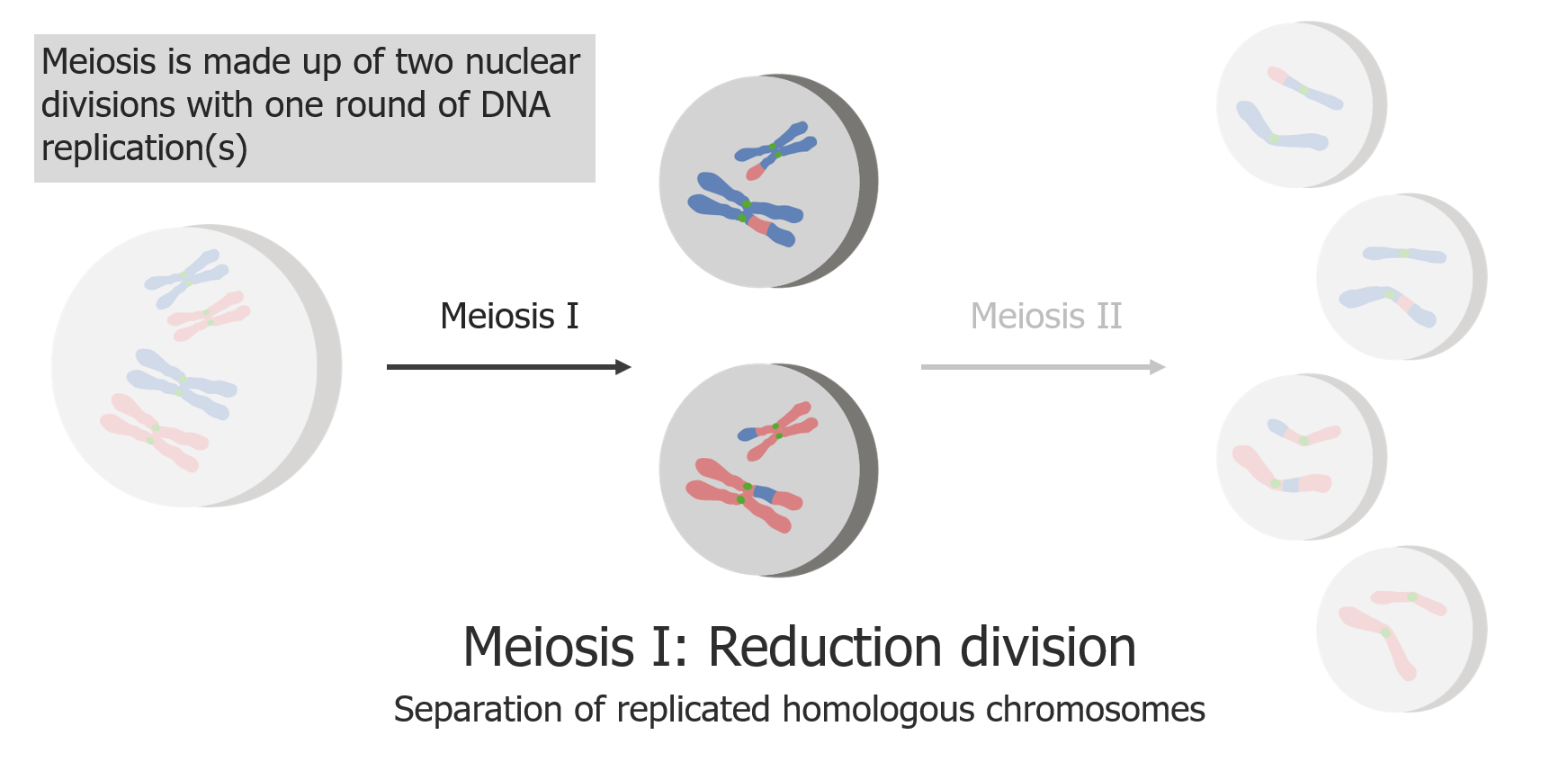
(1) Meiose I é a separação de cromatídeos irmãos replicados.
Imagem por Lecturio.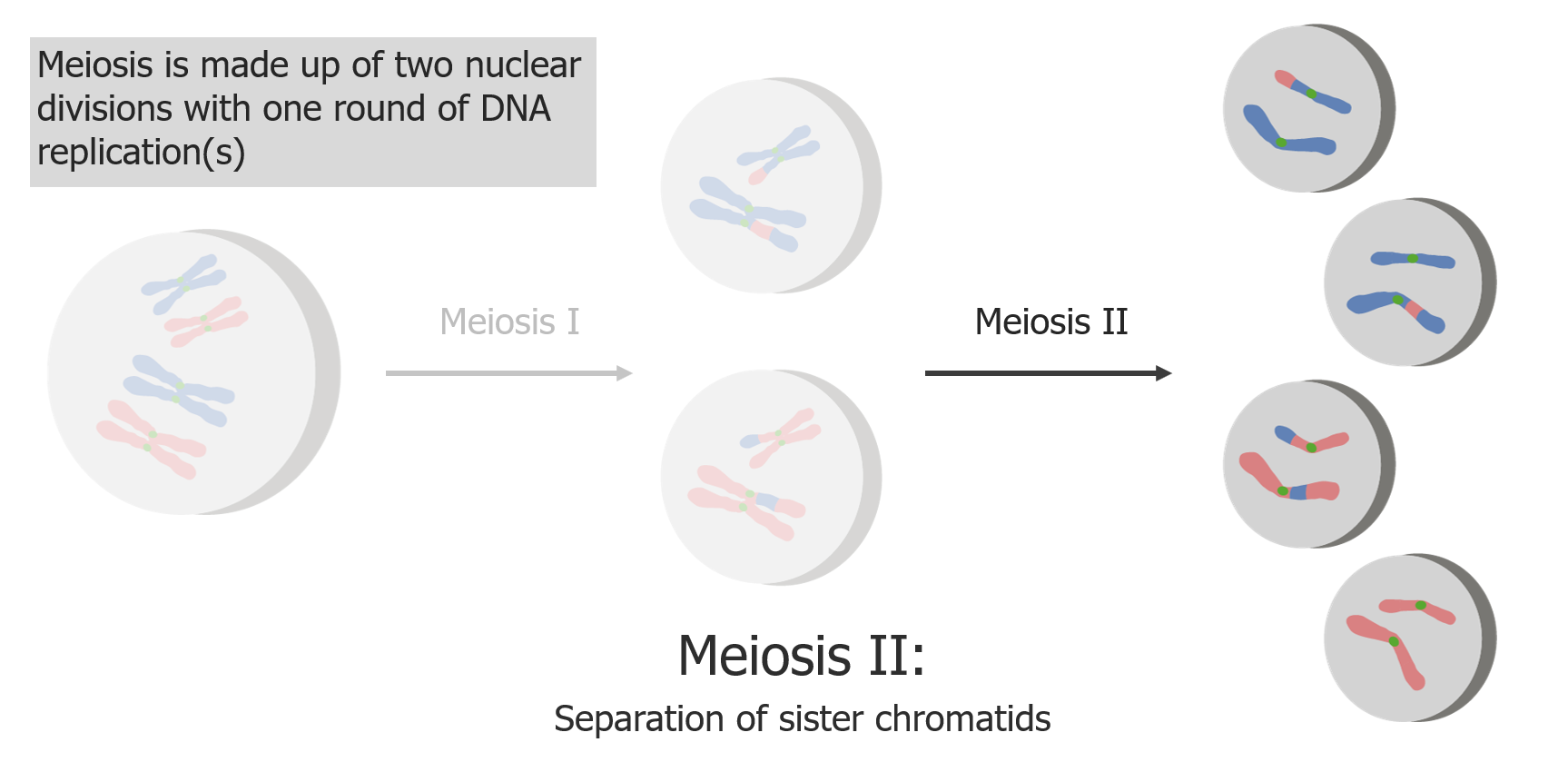
(2) Meiose II resulta na separação de cromatídeos irmãos, produzindo 4 células/gâmetas haplóides.
Imagem por Lecturio.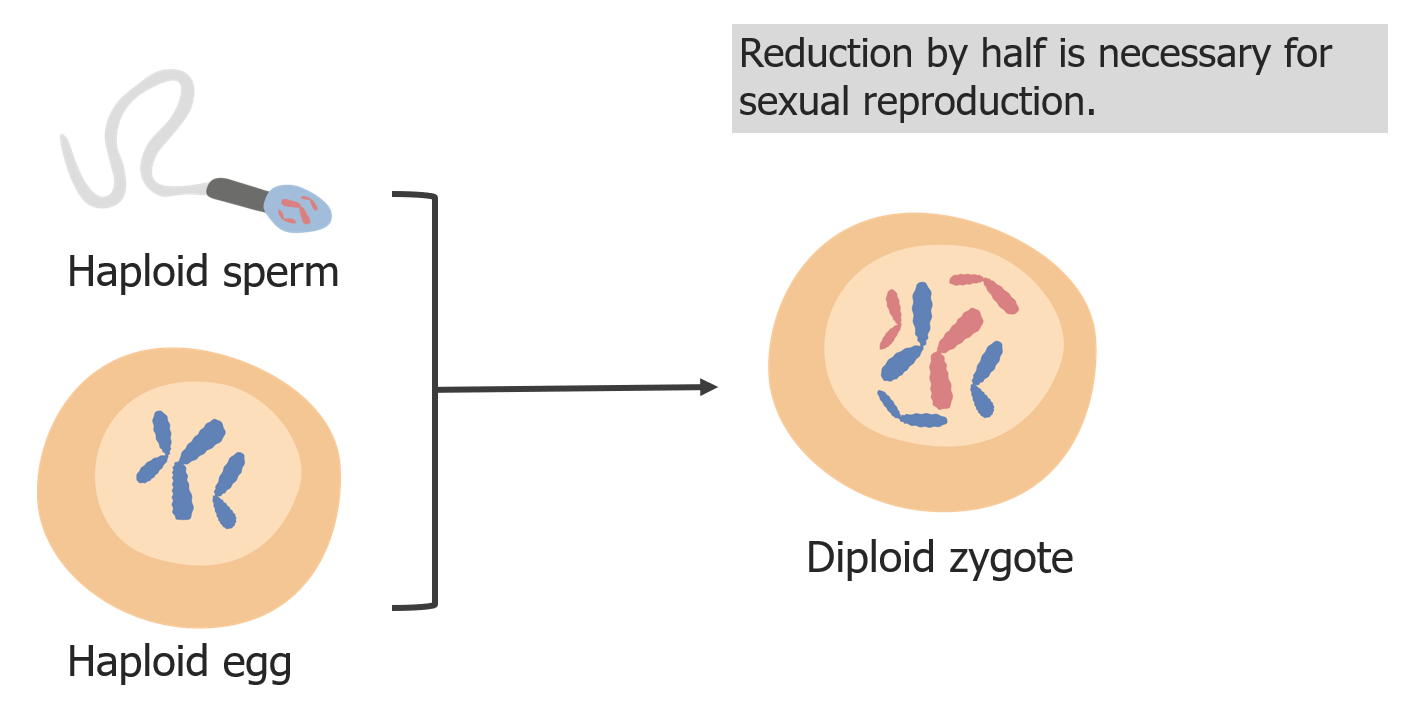
(3) A redução dos cromossomas é necessária para que, na fertilização, o zigoto seja diploide.
Imagem por Lecturio.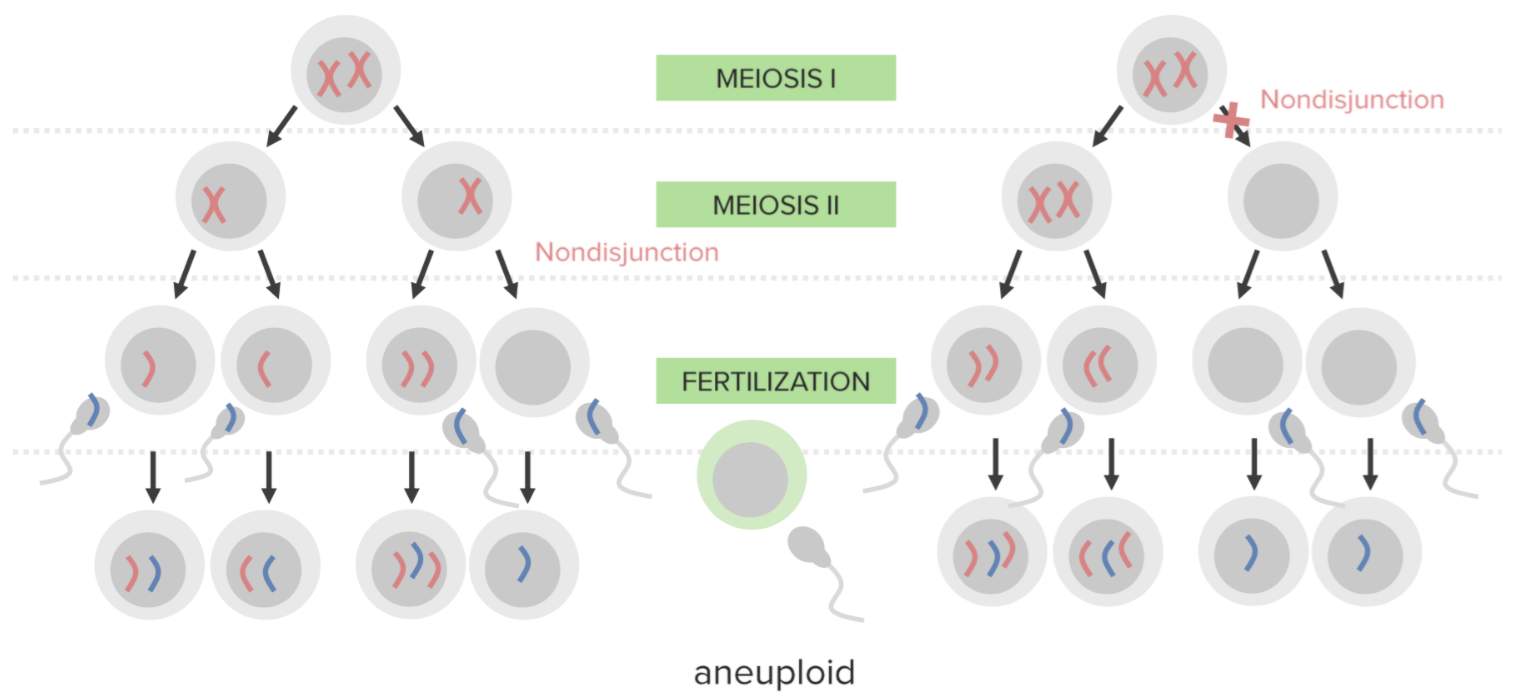
Não disjunção:
Falha na separação adequada de 2 cromossomas homólogos ou dos cromatídeos irmãos durante a divisão celular.
A não disjunção resulta em aneuploidia, um estado de desequilíbrio cromossómico.

Diagrama que explica o mecanismo etiológico da não disjunção nas trissomias:
Um óvulo que carrega 2 cópias do mesmo cromossoma adquire outro quando fertilizado (resultando em 3 cópias).
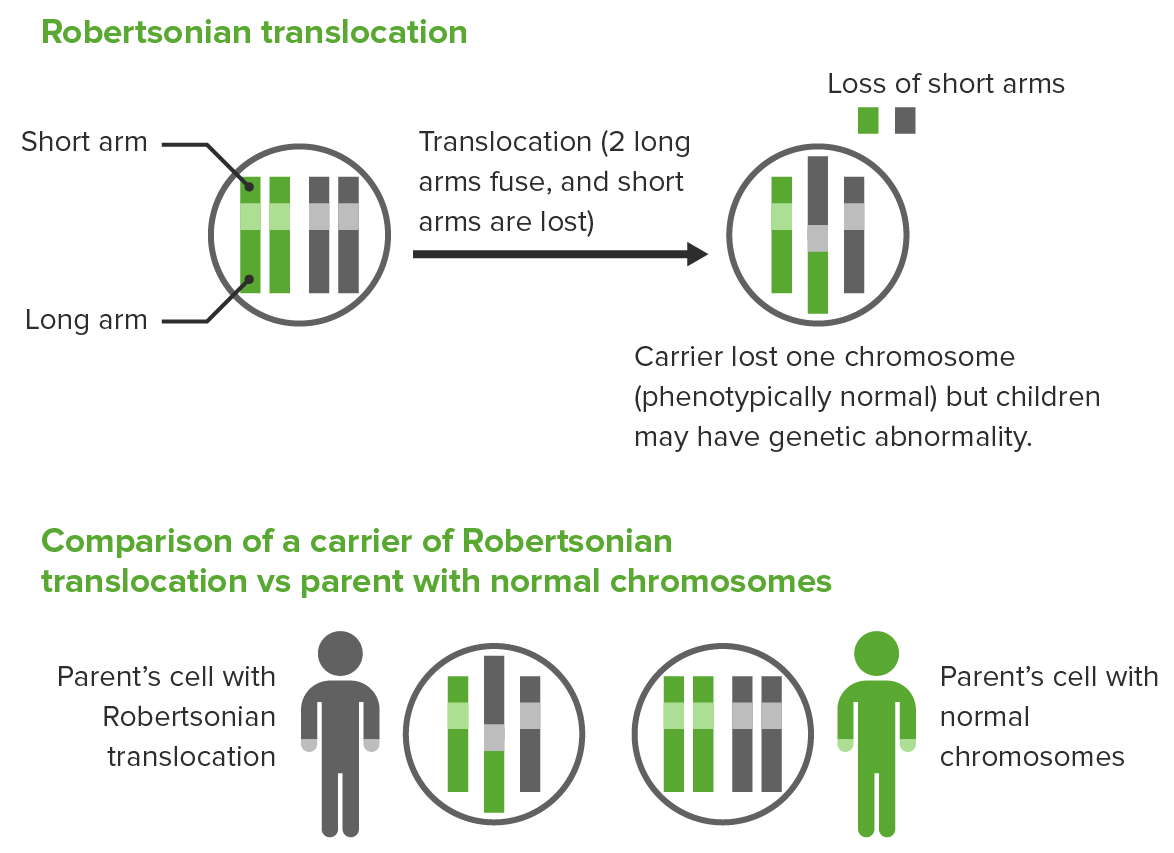
Translocação Robertsoniana:
Os 2 braços longos de diferentes cromossomas acrocêntricos fundem-se, resultando na perda de braços curtos e na perda de 1 cromossoma.
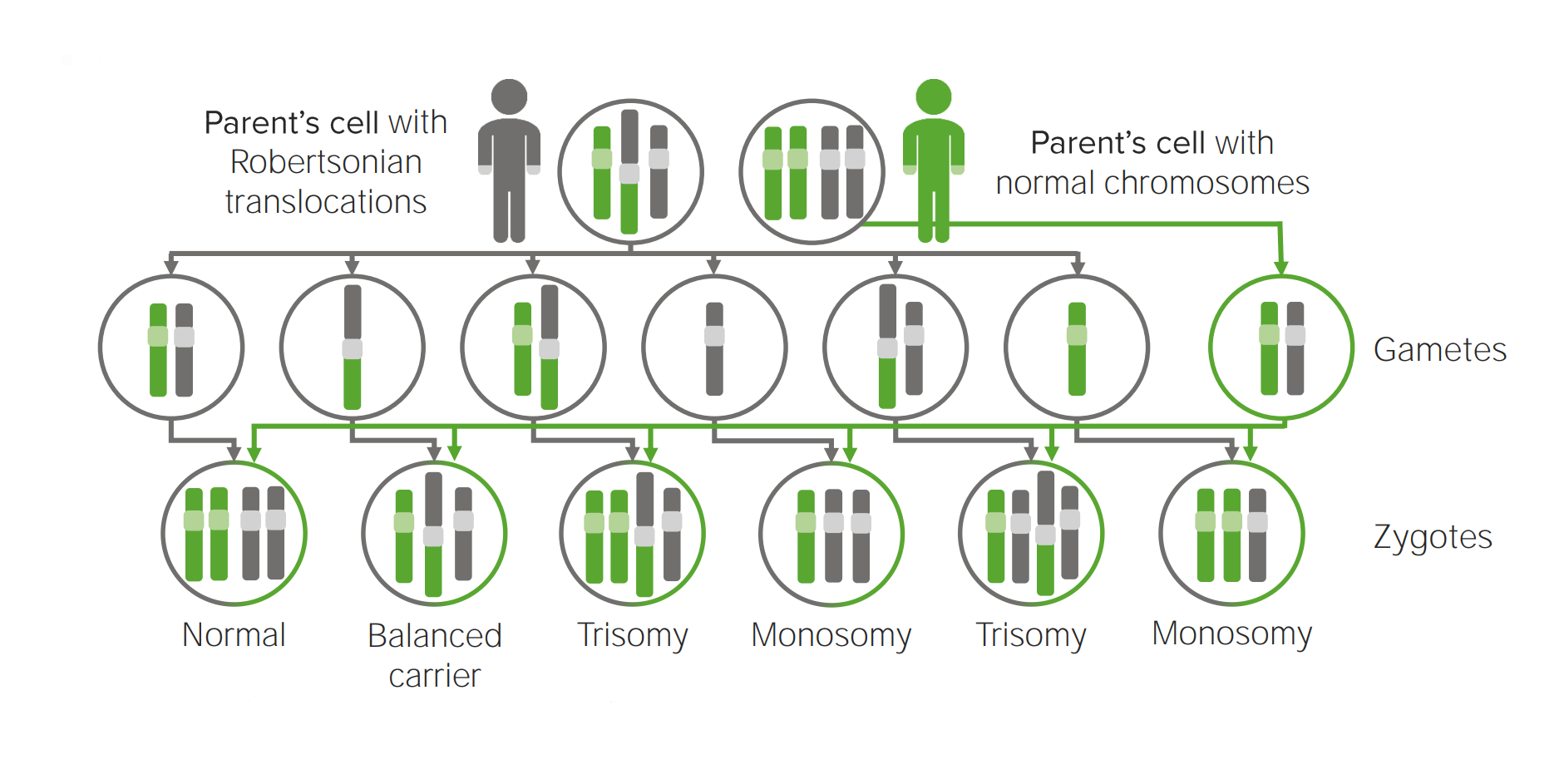
Diagrama que explica o mecanismo etiológico da translocação Robertsoniana em trissomias.
A translocação para outro cromossoma irá possivelmente criar uma célula portadora de 3 cópias do cromossoma implicado.
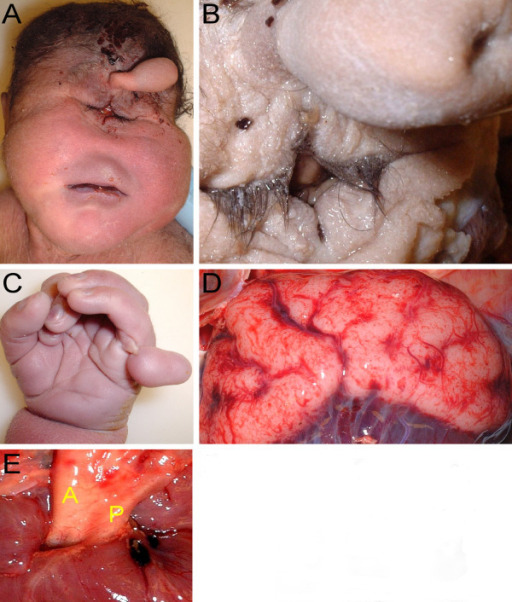
Síndrome de Patau: criança de 37s + 2d do sexo masculino com síndrome de Patau demonstrando holoprosencefalia alobar com ciclopia.
A) As características faciais incluem uma testa inclinada com uma probóscide superior a uma única fissura palpebral central.
B) Grande plano das pálpebras fundidas e probóscide evidenciando uma única narina
C) Polidactilia (seis dígitos)
D) Visão posterior do cérebro mostrando giroses indistintas, fusão dos hemisférios e encefalocelo occipital
E) Transposição da aorta (A) e do tronco pulmonar hipoplásico (P)
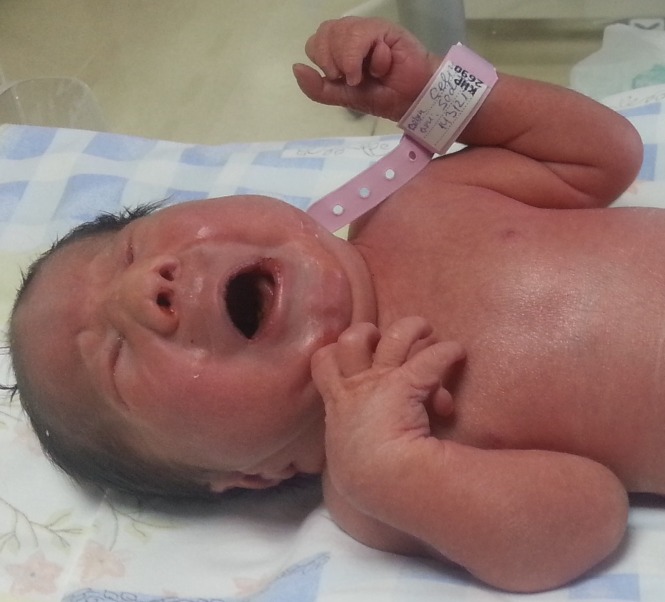
Características da trissomia 13: polidactilia, microftalmia, orelhas baixas e raiz nasal deprimida
Imagem: “Sacrococcygeal teratoma associated with trisomy 13” por Dorum BA, Köksal N, Özkan H, Karakaya S, Akgül AK. Licença: CC BY 3.0| 1º trimestre | 2º trimestre | ||||||
|---|---|---|---|---|---|---|---|
| TN | Proteína A plasmática associada à gravidez | β-hCG | AFP AFP The first alpha-globulins to appear in mammalian sera during fetal development and the dominant serum proteins in early embryonic life. Hepatocellular Carcinoma (HCC) and Liver Metastases | Estriol Estriol A hydroxylated metabolite of estradiol or estrogen that has a hydroxyl group at C3, 16-alpha, and 17-beta position. Estriol is a major urinary estrogen. During pregnancy, a large amount of estriol is produced by the placenta. Isomers with inversion of the hydroxyl group or groups are called epiestriol. Noncontraceptive Estrogen and Progestins | β-hCG | Inhibina A | |
| Trissomia 13 | ↑ | ↓↓ | ↓ | Inalterado | Inalterado | Inalterado | Inalterado |
| Trissomia 18 | ↑↑ | ↓↓ | ↓↓ | ↓ | ↓↓ | ↓↓ | Inalterado |
| Trissomia 21 | ↑↑ | ↓↓ | ↑ | ↓ | ↓ | ↑ | ↑ |
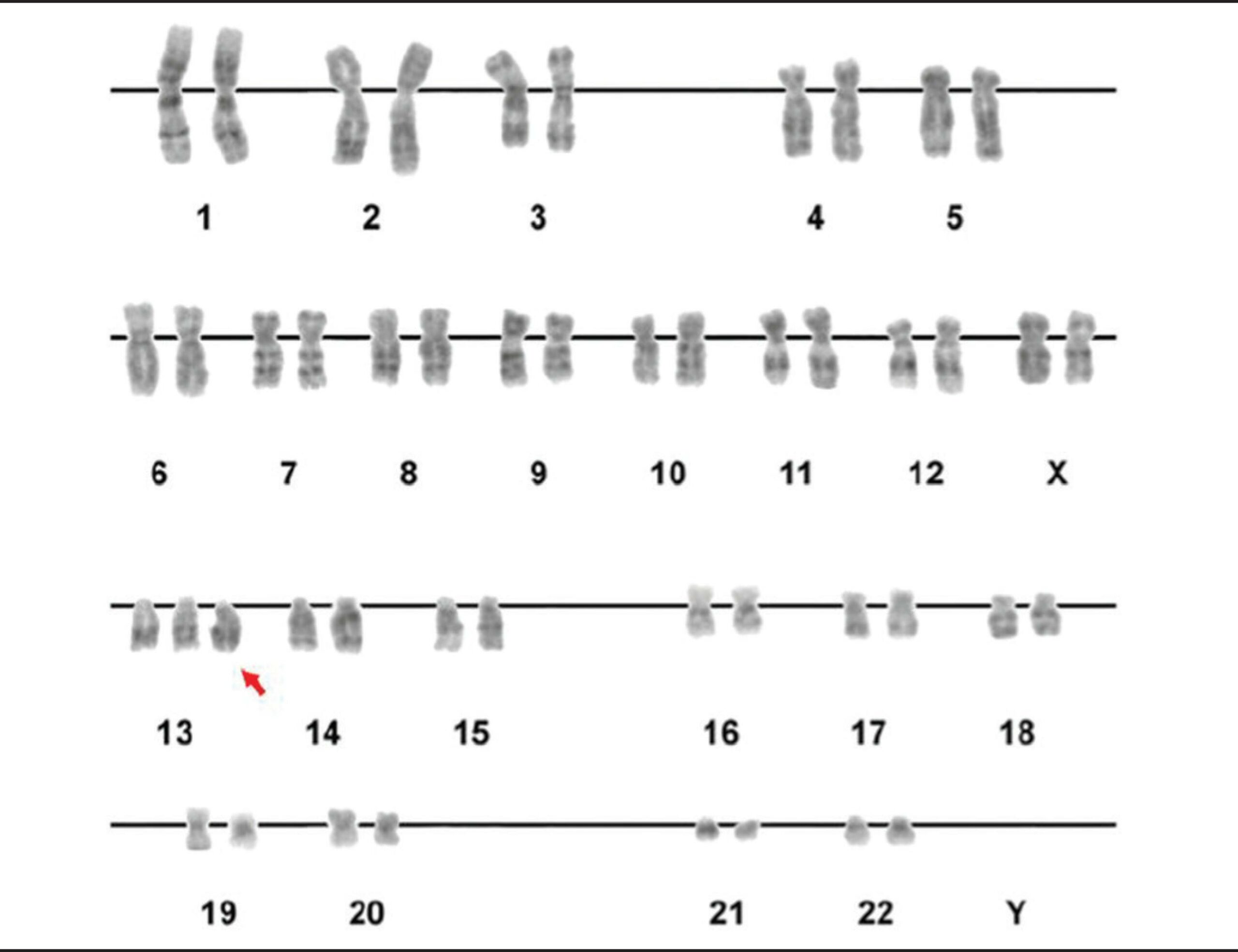
Um cariótipo que mostra a trissomia do 13.º cromossoma (seta vermelha)
Imagem: “Figure 2” por Luiza E. Dorfman, et al. Licença: CC BY 4.0