La trisomía 13, o síndrome de Patau, es un síndrome genético causado por la presencia de 3 copias del cromosoma 13. Siendo la 3era trisomía más común, el síndrome de Patau tiene una incidencia de 1 por cada 10 000 nacidos vivos y es más común en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las mujeres. La mayoría de los LOS Neisseria casos de síndrome de Patau se diagnostican de forma prenatal mediante tamizaje materno y ultrasonido. Más de la mitad de los LOS Neisseria embarazos terminan en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum abortos espontáneos. Si el embarazo llega a término, se recomienda que un centro especializado se encargue del parto y la atención neonatal. En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el neonato, los LOS Neisseria hallazgos incluyen malformaciones craneofaciales y cardíacas, discapacidad intelectual grave y una esperanza de vida muy reducida. La mayoría de los LOS Neisseria bebés no sobreviven más allá de los LOS Neisseria 3 meses. Sin tratamiento disponible y con una esperanza de supervivencia nula, la familia recibe apoyo y recursos para navegar a través del curso natural de la enfermedad.
Last updated: Dec 15, 2025
La trisomía 13 o síndrome de Patau se define por la presencia de 3 copias del cromosoma 13.
“A los 13 años se entra en la Pubertad”: P como en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el síndrome de Patau o 13 para la Trisomía 13
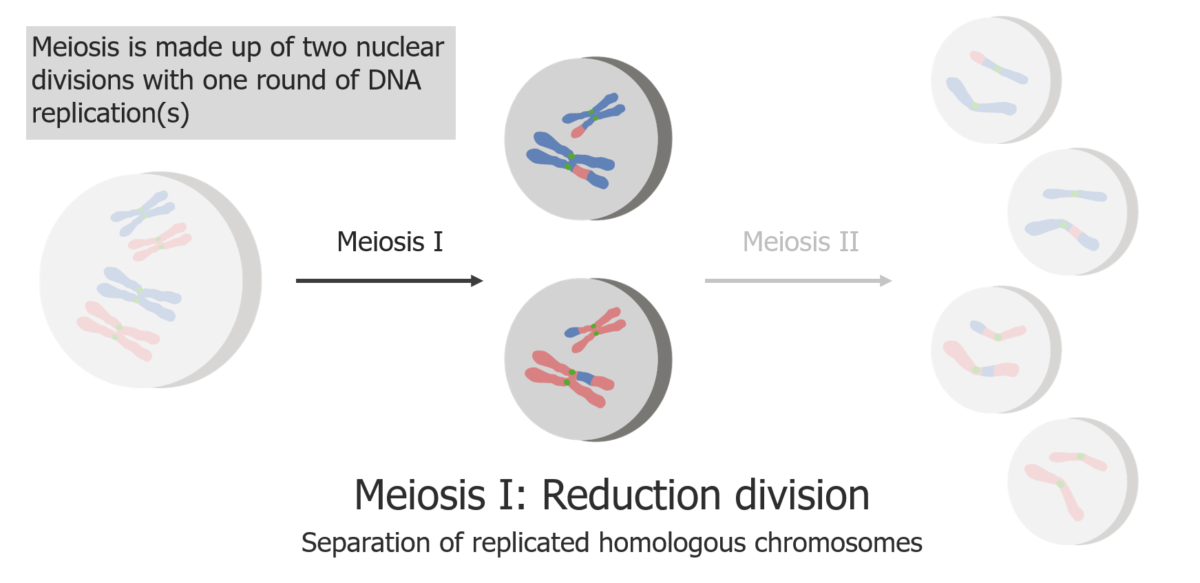
(1) La meiosis I es la separación de las cromátidas hermanas replicadas.
Imagen por Lecturio.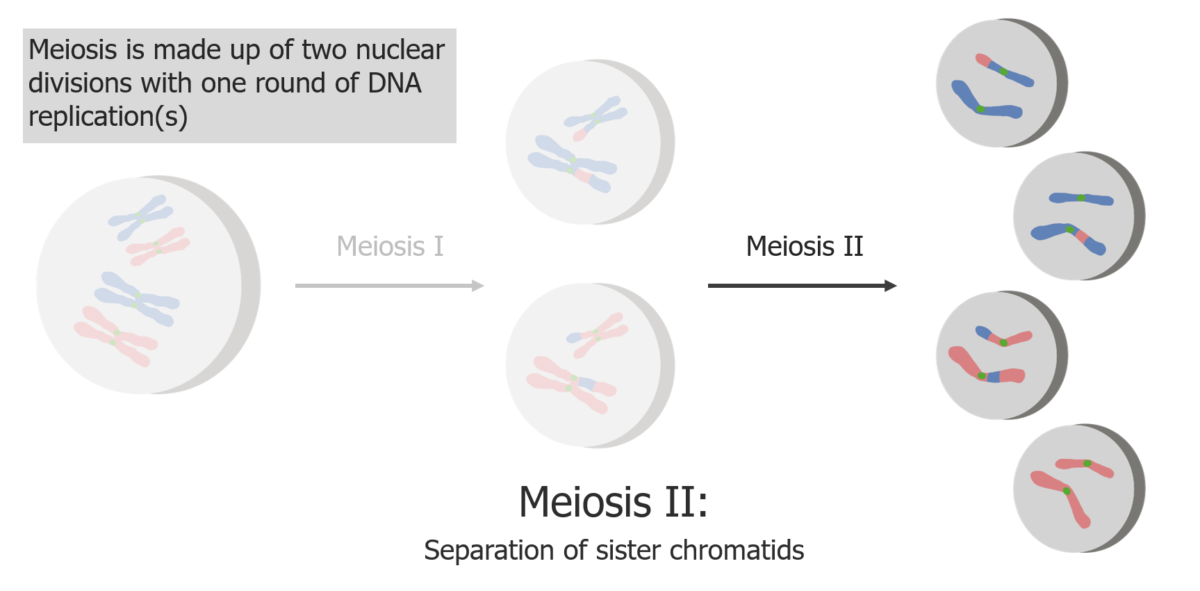
(2) La meiosis II da lugar a la separación de las cromátidas hermanas, produciendo 4 células/gametos haploides.
Imagen por Lecturio.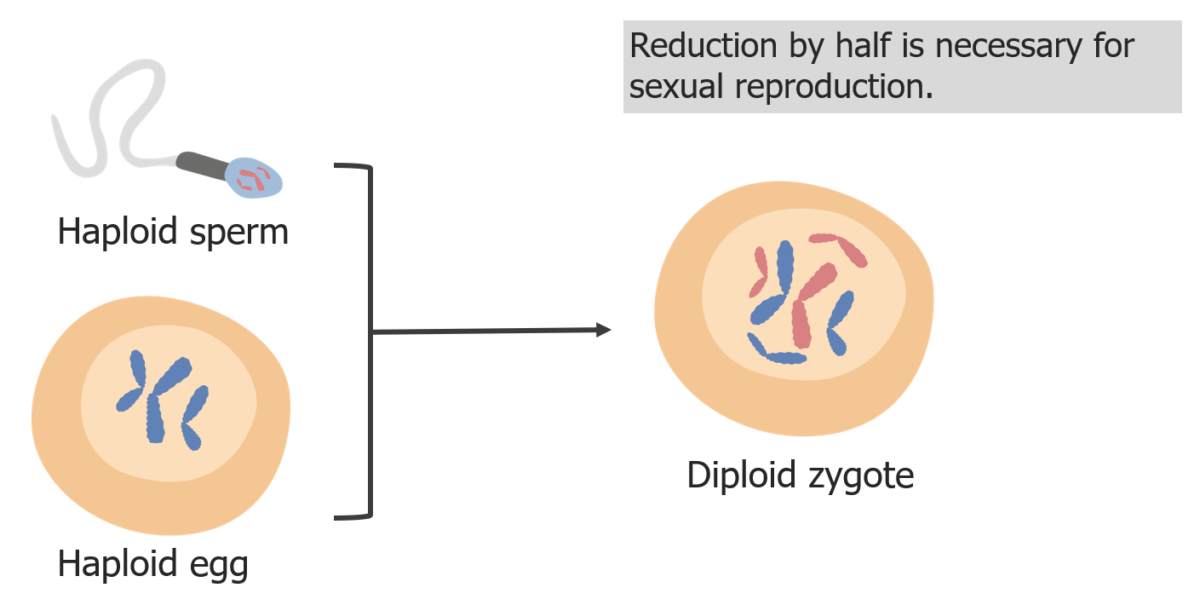
(3) La reducción de los cromosomas es necesaria para que en la fecundación el cigoto sea diploide.
Imagen por Lecturio.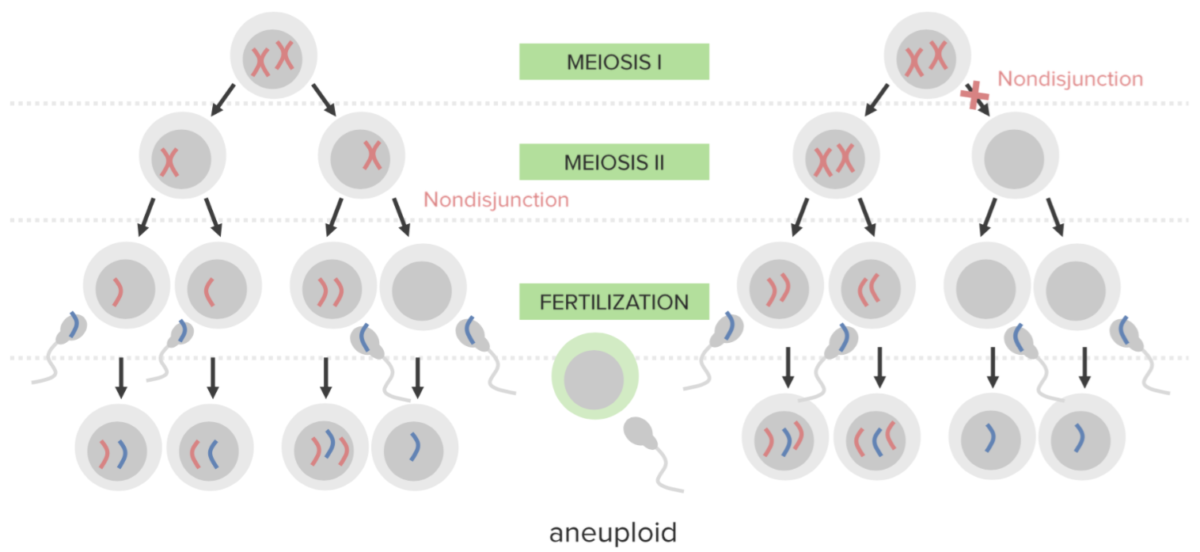
La no disyunción:
Fallo en la separación adecuada de 2 cromosomas homólogos o de las cromátidas hermanas durante la división celular.
La no disyunción da lugar a la aneuploidía, un estado de desequilibrio cromosómico.
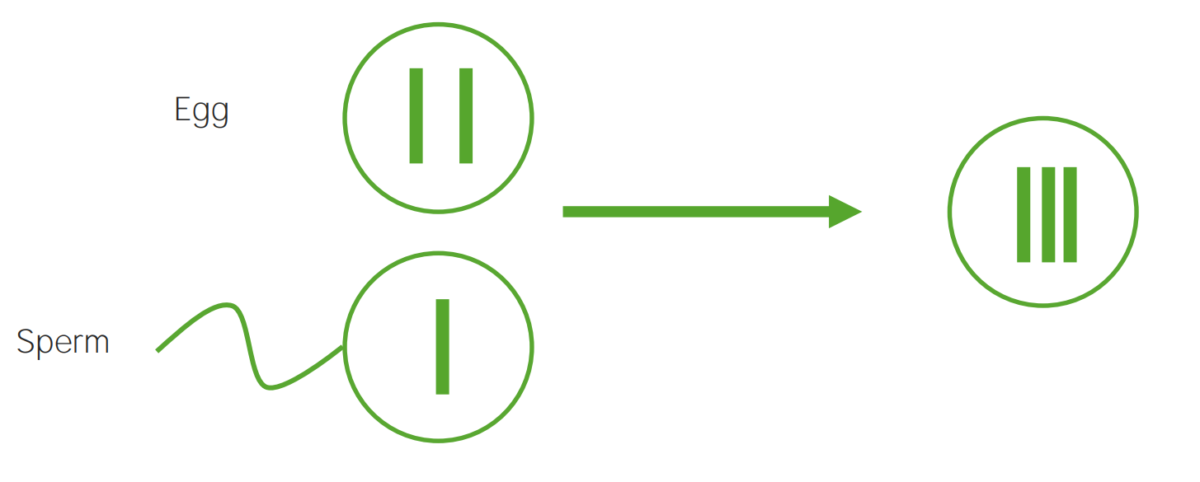
Diagrama que explica el mecanismo etiológico de la no disyunción en las trisomías:
Un óvulo que lleva 2 copias del mismo cromosoma adquiere otro al ser fecundado (dando lugar a 3 copias).
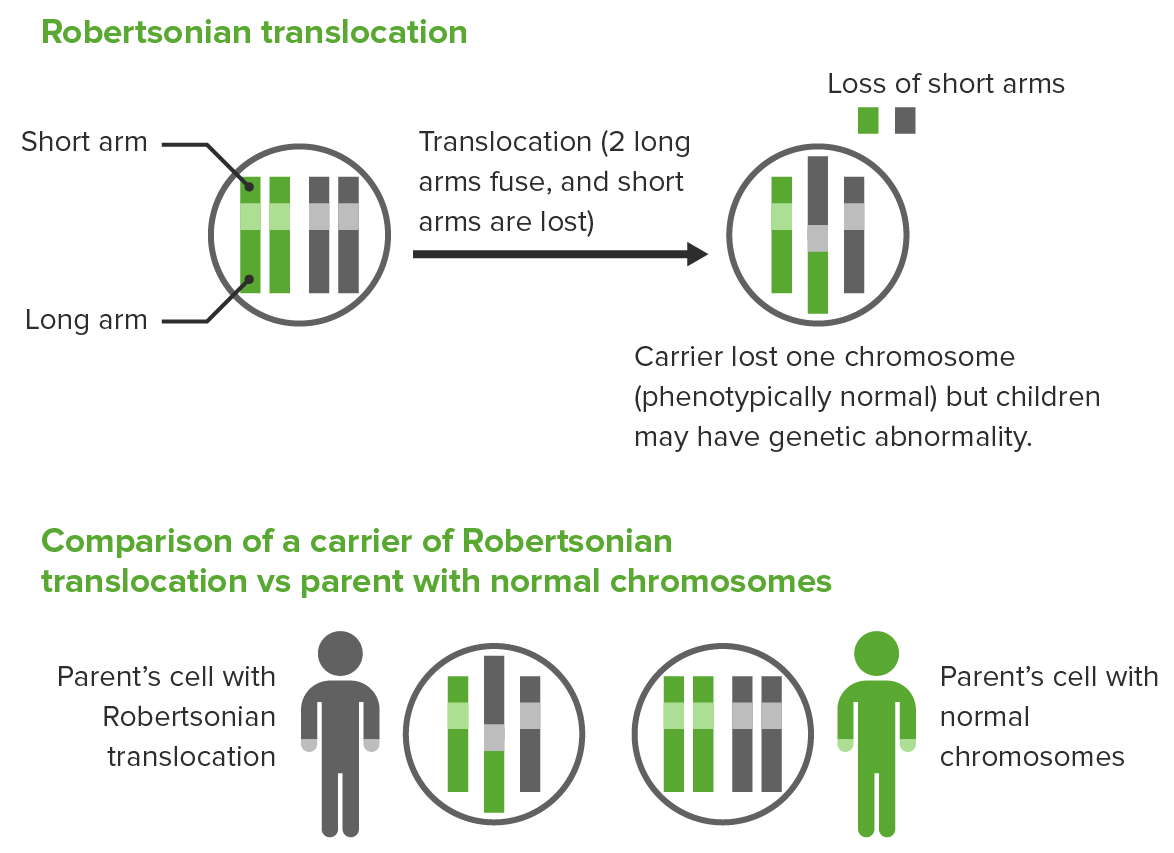
Translocación robertsoniana:
Los 2 brazos largos de diferentes cromosomas acrocéntricos se fusionan, dando lugar a la pérdida de los brazos cortos y a la pérdida de 1 cromosoma.
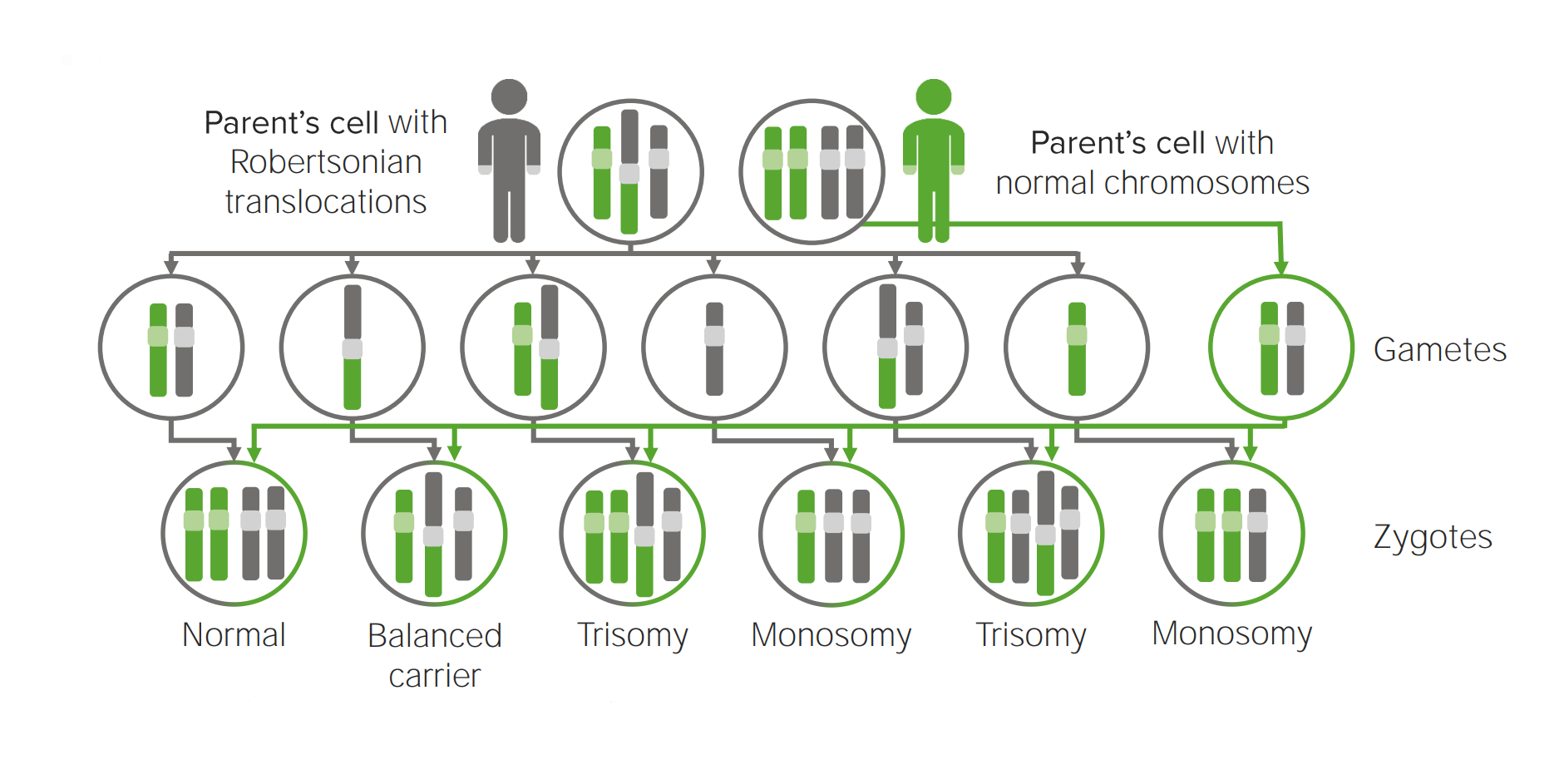
Diagrama que explica el mecanismo etiológico de la translocación robertsoniana en las trisomías.
La translocación a otro cromosoma creará posiblemente una célula portadora de 3 copias del cromosoma implicado.
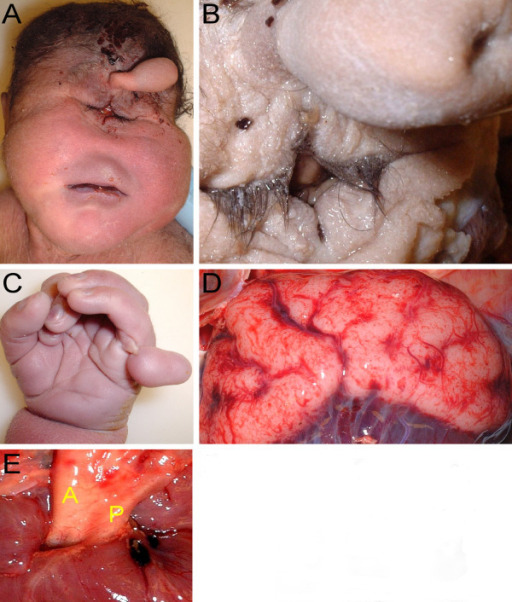
Síndrome de Patau: niño varón de 37 2/7 semanas de edad gestacional con síndrome de Patau que presenta holoprosencefalia alobar con ciclopía.
A) Los rasgos faciales incluyen una frente inclinada con una probóscide superior a una única fisura palpebral central.
B) Primer plano de los párpados fusionados y la probóscide mostrando una sola fosa nasal
C) Polidactilia con seis dedos
D) Vista posterior del cerebro mostrando giros indistintos, fusión de los hemisferios y encefalocele occipital
E) Transposición de aorta (A) y tronco pulmonar hipoplásico (P)
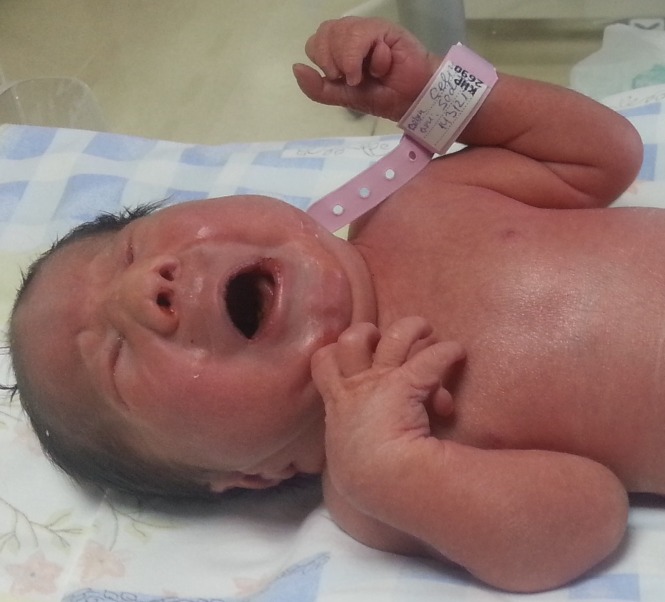
Características de la trisomía 13: polidactilia, microftalmia, orejas de implantación baja y puente nasal deprimido
Imagen: “Sacrococcygeal teratoma associated with trisomy 13” por Dorum BA, Köksal N, Özkan H, Karakaya S, Akgül AK. Licencia: CC BY 3.0| 1er trimestre | 2do trimestre | ||||||
|---|---|---|---|---|---|---|---|
| TNF TNF Tumor necrosis factor (TNF) is a major cytokine, released primarily by macrophages in response to stimuli. The presence of microbial products and dead cells and injury are among the stimulating factors. This protein belongs to the TNF superfamily, a group of ligands and receptors performing functions in inflammatory response, morphogenesis, and cell proliferation. Tumor Necrosis Factor (TNF) | Proteína plasmática A asociada al AL Amyloidosis embarazo | β-hCG | AFP AFP The first alpha-globulins to appear in mammalian sera during fetal development and the dominant serum proteins in early embryonic life. Hepatocellular Carcinoma (HCC) and Liver Metastases | Estriol Estriol A hydroxylated metabolite of estradiol or estrogen that has a hydroxyl group at C3, 16-alpha, and 17-beta position. Estriol is a major urinary estrogen. During pregnancy, a large amount of estriol is produced by the placenta. Isomers with inversion of the hydroxyl group or groups are called epiestriol. Noncontraceptive Estrogen and Progestins | β-hCG | Inhibina A | |
| Trisomía 13 | ↑ | ↓↓ | ↓ | Sin cambios | Sin cambios | Sin cambios | Sin cambios |
| Trisomía 18 | ↑↑ | ↓↓ | ↓↓ | ↓ | ↓↓ | ↓↓ | Sin cambios |
| Trisomía 21 | ↑↑ | ↓↓ | ↑ | ↓ | ↓ | ↑ | ↑ |
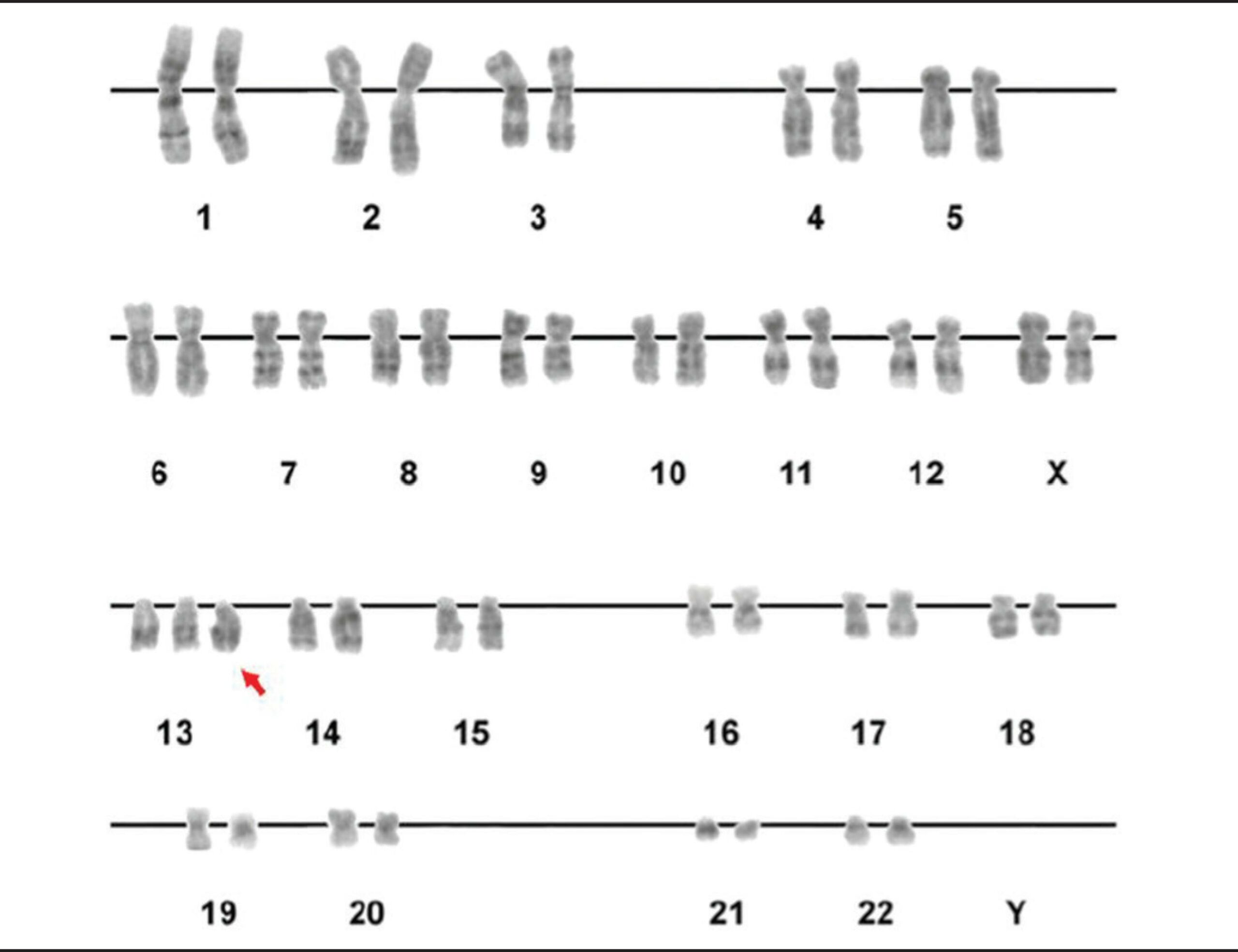
Un cariotipo que muestra la trisomía del cromosoma 13 (flecha roja)
Imagen: “Figure 2” por Luiza E. Dorfman, et al. Licencia: CC BY 4.0