


A síndrome de Kallmann ( SK SK Seborrheic keratosis (sk) is the most common benign epithelial cutaneous neoplasm. The condition consists of immature keratinocytes. Seborrheic keratosis is the most common benign skin tumor in middle-aged and elderly adults and presents as a sharply demarcated, exophytic, skin lesion that may be tan or black and has a "stuck-on" appearance. Seborrheic Keratosis), também denominada síndrome olfato-genital, é uma doença genética que causa hipogonadismo hipogonadotrófico devido à diminuição da secreção da hormona libertadora de gonadotrofina (GnRH) pelo hipotálamo. Ambos os sexos podem ser afetados, embora a incidência seja muito maior no sexo masculino. A falta de hormonas sexuais resulta no compromisso do desenvolvimento pubertário. Caracteristicamente, existe uma ausência ou diminuição do olfato (hiposmia ou anosmia Anosmia Complete or severe loss of the subjective sense of smell. Loss of smell may be caused by many factors such as a cold, allergy, olfactory nerve diseases, viral respiratory tract infections (e.g., COVID-19), aging and various neurological disorders (e.g., Alzheimer disease). Cranial Nerve Palsies) associada, o que ajuda a diferenciar a SK SK Seborrheic keratosis (sk) is the most common benign epithelial cutaneous neoplasm. The condition consists of immature keratinocytes. Seborrheic keratosis is the most common benign skin tumor in middle-aged and elderly adults and presents as a sharply demarcated, exophytic, skin lesion that may be tan or black and has a "stuck-on" appearance. Seborrheic Keratosis de outras doenças. O diagnóstico é feito pelos níveis hormonais séricos e exames de imagem cerebral que mostram a ausência de estruturas olfativas. Os testes Testes Gonadal Hormones genéticos podem ajudar a estabelecer um diagnóstico definitivo. O tratamento consiste em terapia de reposição hormonal.
Last updated: Dec 15, 2025
Os genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure listados acima estão envolvidos na migração de neurónios olfativos e neurónios envolvidos na produção do hormona libertadora de gonadotrofina (GnRH) para o seu local funcional adequado no cérebro durante o desenvolvimento.
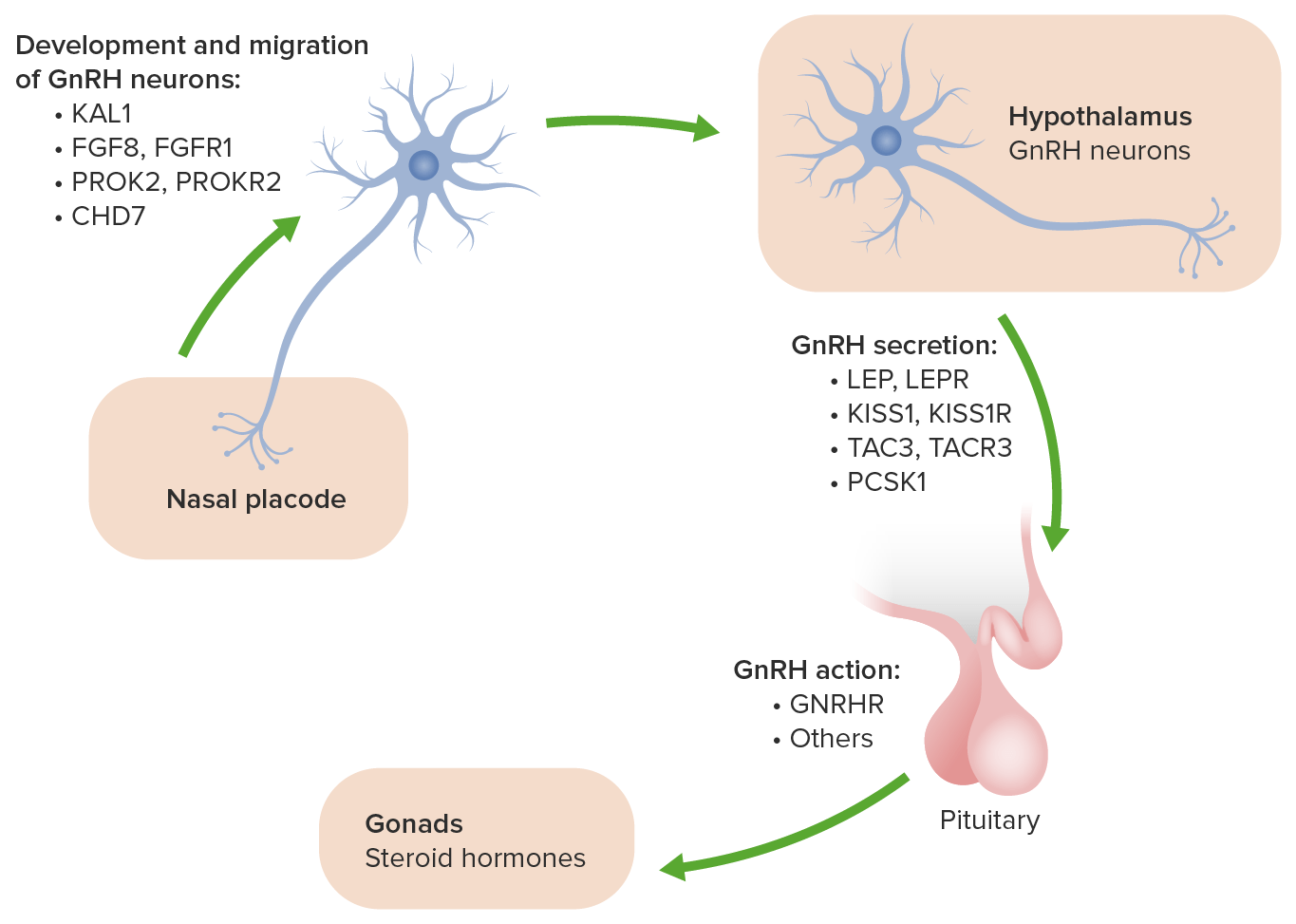
A base genética e molecular do hipogonadismo hipogonadotrófico idiopático :
O gene ANOS1 era previamento chamado KAL1. Causa a forma da síndrome de Kallmann ligada ao X e está associado aos sintomas adicionais de anosmia, sincinésia bimanual e agenesia renal.
A apresentação clínica é altamente variável devido às diferentes mutações genéticas e níveis de penetrância. No entanto, todos os indivíduos com SK SK Seborrheic keratosis (sk) is the most common benign epithelial cutaneous neoplasm. The condition consists of immature keratinocytes. Seborrheic keratosis is the most common benign skin tumor in middle-aged and elderly adults and presents as a sharply demarcated, exophytic, skin lesion that may be tan or black and has a “stuck-on” appearance. Seborrheic Keratosis apresentam desenvolvimento sexual anormal e hiposmia ou anosmia Anosmia Complete or severe loss of the subjective sense of smell. Loss of smell may be caused by many factors such as a cold, allergy, olfactory nerve diseases, viral respiratory tract infections (e.g., COVID-19), aging and various neurological disorders (e.g., Alzheimer disease). Cranial Nerve Palsies.
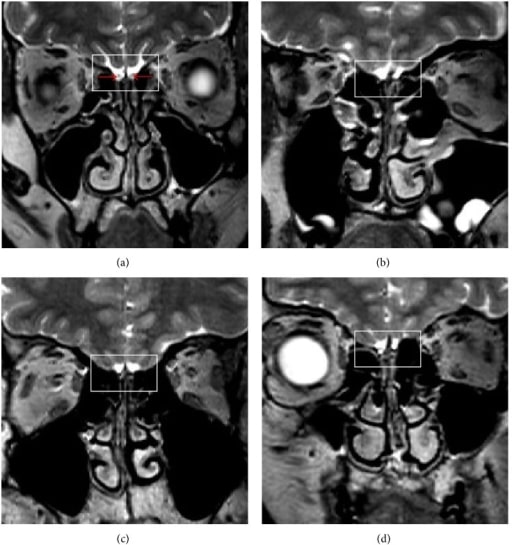
Achados de ressonância magnética do cérebro na SK
Imagem por ressonância magnética (RM) de 3 pacientes do sexo masculino com SK (b) – (d). A imagem (a) mostra estruturas normais num controlo saudável. As setas vermelhas indicam bulbos olfativos. As imagens (b), (c) e (d) mostram a ausência do bulbo olfativo bilateral, do trato olfativo e do sulco (quadrados), respetivamente.
