El síndrome de Kallmann, también llamado síndrome olfativo-genital, es una afección genética que causa hipogonadismo hipogonadotrópico debido a la disminución de la secreción de la hormona liberadora de gonadotropina (GnRH, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) por el hipotálamo. Ambos sexos pueden verse afectados, aunque la incidencia es mucho mayor en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria varones. La falta de hormonas sexuales provoca una alteración del desarrollo puberal. Es característico que haya una ausencia o disminución del sentido del olfato (hiposmia o anosmia Anosmia Complete or severe loss of the subjective sense of smell. Loss of smell may be caused by many factors such as a cold, allergy, olfactory nerve diseases, viral respiratory tract infections (e.g., COVID-19), aging and various neurological disorders (e.g., Alzheimer disease). Cranial Nerve Palsies), lo que ayuda a diferenciar el síndrome de Kallmann de otras enfermedades. El diagnóstico se realiza mediante los LOS Neisseria niveles hormonales en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum sangre y la imagenología cerebral que muestran la ausencia de estructuras olfativas. Las pruebas genéticas pueden ayudar a establecer un diagnóstico definitivo. El tratamiento consiste en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum una terapia de sustitución hormonal.
Last updated: Dec 15, 2025
Los LOS Neisseria genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure mencionados anteriormente están implicados en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la migración de las neuronas olfativas y de las neuronas implicadas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la producción de la hormona liberadora de gonadotropina (GnRH, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) a su lugar funcional adecuado en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el cerebro durante el desarrollo.
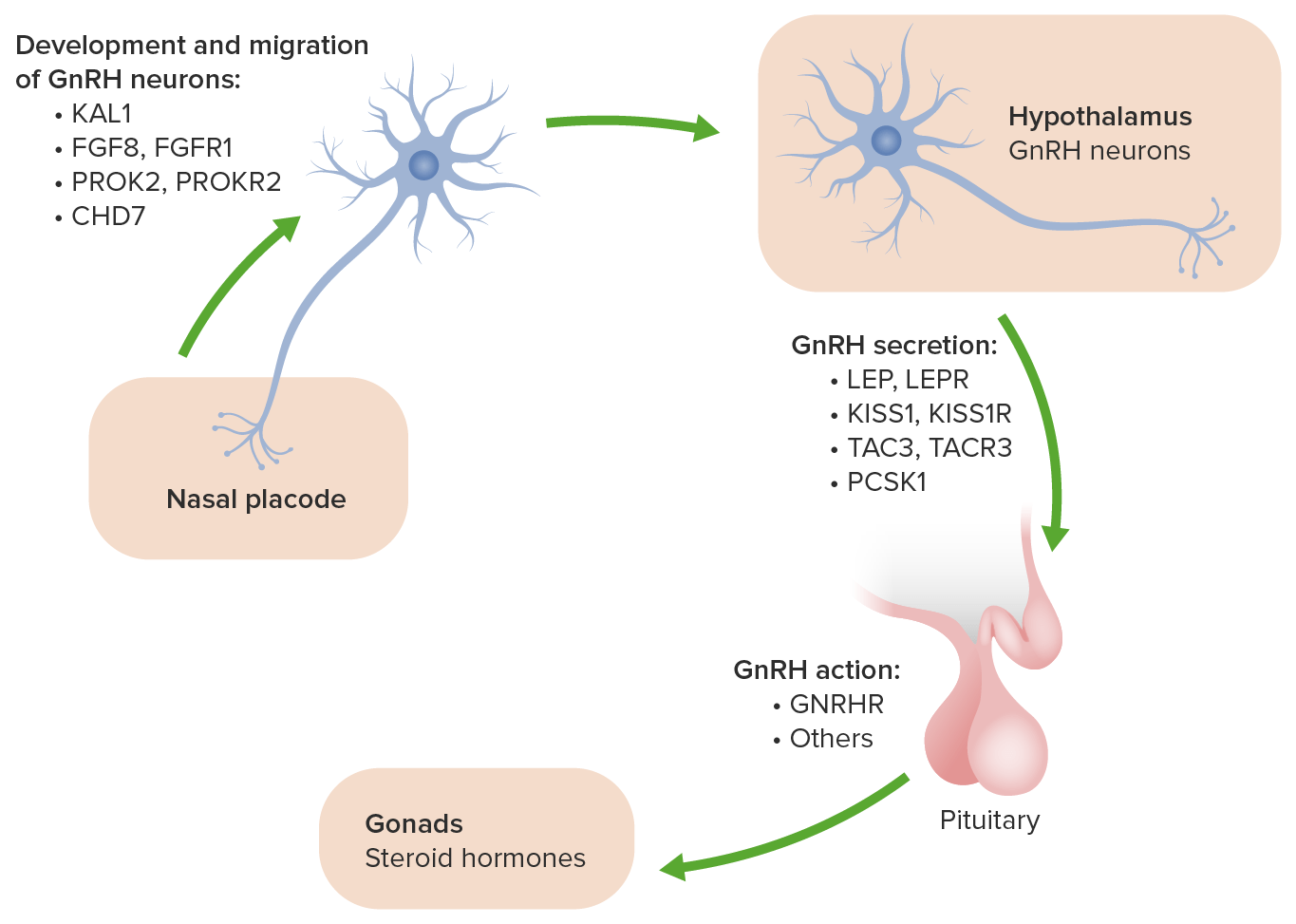
Las bases genéticas y moleculares del hipogonadismo hipogonadotrópico idiopático:
El gen ANOS1 se denominaba anteriormente KAL1. Causa la forma ligada al cromosoma X del síndrome de Kallmann y se asocia con los síntomas adicionales de anosmia, sinquinesis bimanual y agenesia renal.
La presentación clínica es muy variable debido a las diferentes mutaciones genéticas y niveles de penetración. Sin embargo, todos los individuos con síndrome de Kallmann muestran un desarrollo sexual anormal e hiposmia o anosmia Anosmia Complete or severe loss of the subjective sense of smell. Loss of smell may be caused by many factors such as a cold, allergy, olfactory nerve diseases, viral respiratory tract infections (e.g., COVID-19), aging and various neurological disorders (e.g., Alzheimer disease). Cranial Nerve Palsies.
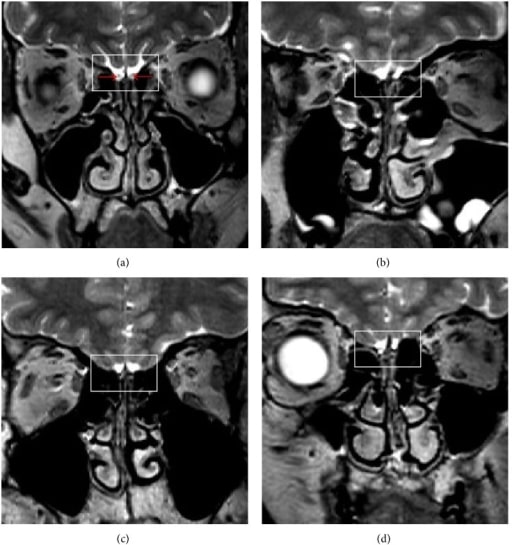
Hallazgos de la RM cerebral en el síndrome de Kallmann
Resonancia magnética (RM) de 3 pacientes varones con síndrome de Kallmann (b)-(d). La imagen (a) muestra estructuras normales en un control sano. Las flechas rojas indican los bulbos olfatorios. Las imágenes (b), (c) y (d) muestran la ausencia del bulbo olfatorio bilateral, el tracto olfatorio y el surco (cuadrados), respectivamente.