


O síndrome de Edwards, ou trissomia 18, é um síndrome genético causado pela presença de um cromossoma 18 extra. O cromossoma extra é ocorre devido a 3 cópias completas do cromossoma 18 ou de um segmento adicional do cromossoma 18. É a segunda trissomia mais comum, vista em 1 em cada 5.500 nados-vivos e aumenta com a idade materna. Muitos casos são detetados na fase pré-natal, com rastreio materno e resultados de ecografias. As anormalidades incluem restrição de crescimento intrauterino (RCIU), dedos sobrepostos, características crânio-faciais típicas, pés virados externamente e com o calcanhar saliente ("Rocker-bottom feet") e defeitos cardíacos congénitos. A trissomia 18 resulta frequentemente em aborto. Nas gravidezes de termo, a maioria das mortes ocorre durante os primeiros 6 meses de vida. O parto num centro especializado é recomendado para gravidezes de termo e a intervenção é baseada nas anormalidades associadas.
Last updated: Dec 15, 2025
O síndrome de Edwards, ou trissomia 18, é definido pela presença de 3 cópias do cromossoma 18.
Mnemónica para o síndrome de Edwards (trissomia 18): “Aos 18 anos, podes votar nas eleições.”
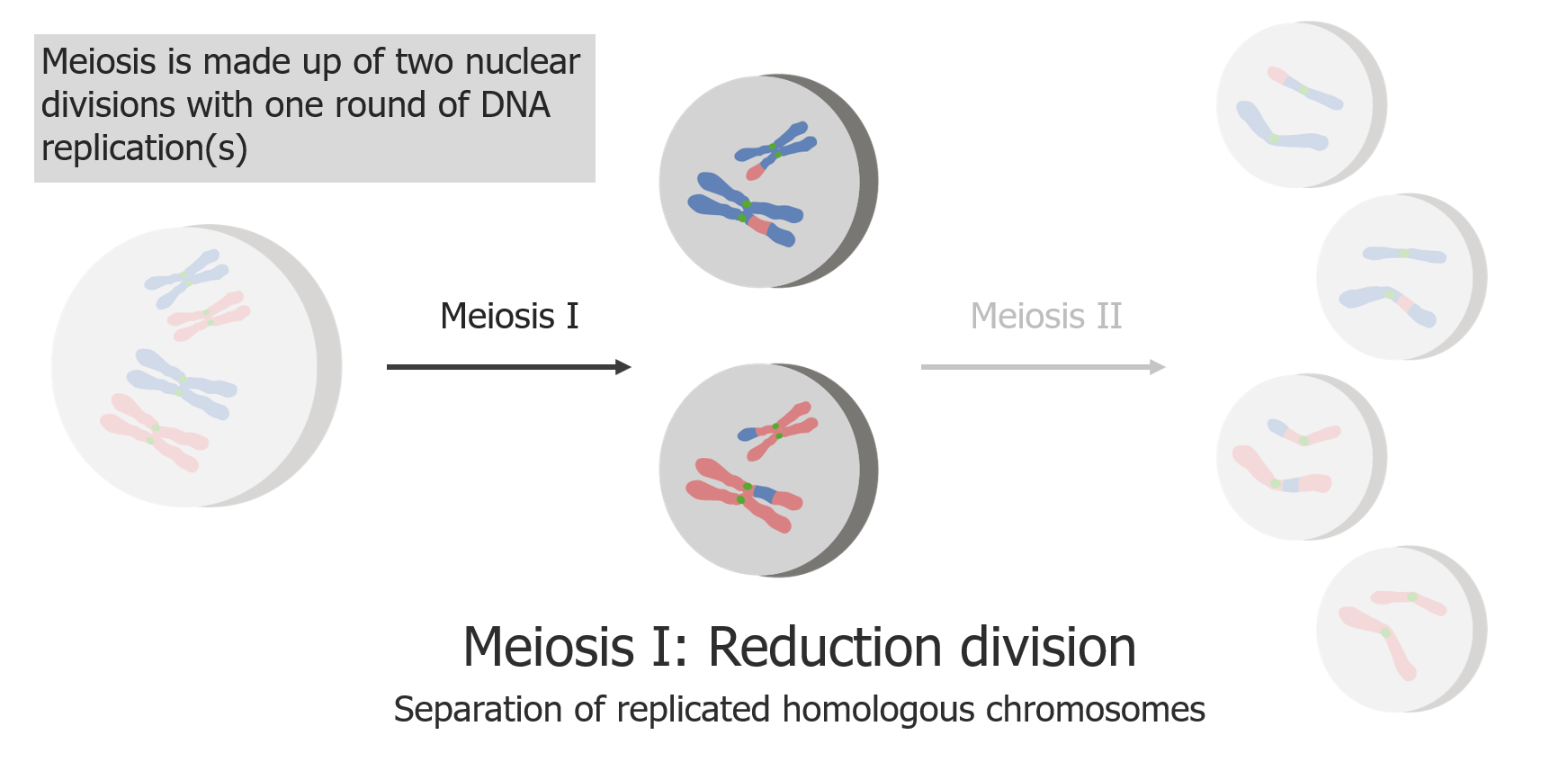
Meiose I é uma separação de cromatídeos irmãos replicados.
Imagem por Lecturio.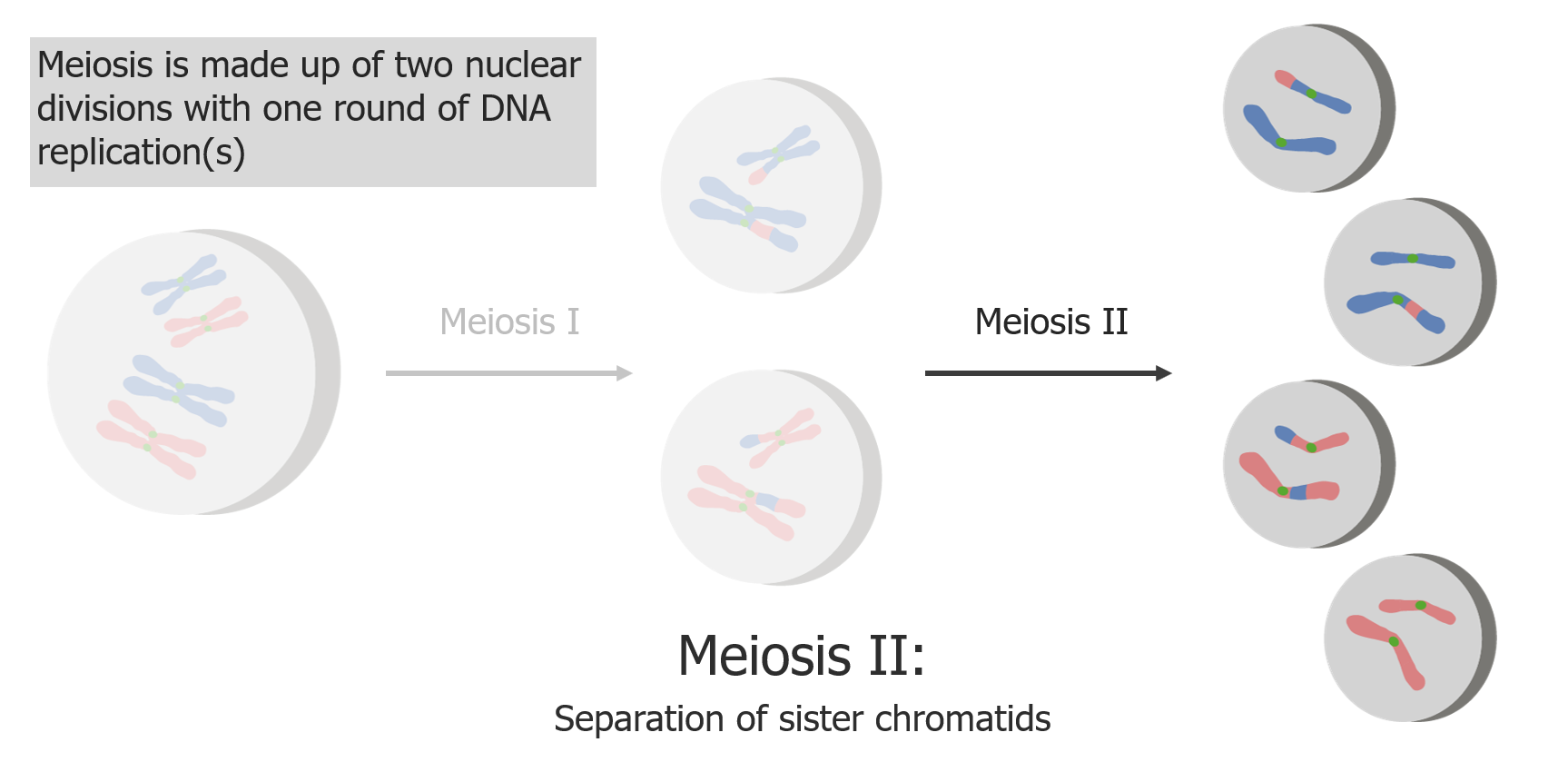
Meiose II é uma separação de cromatídeos irmãos, produzindo 4 células/gâmetas haplóides.
Imagem por Lecturio.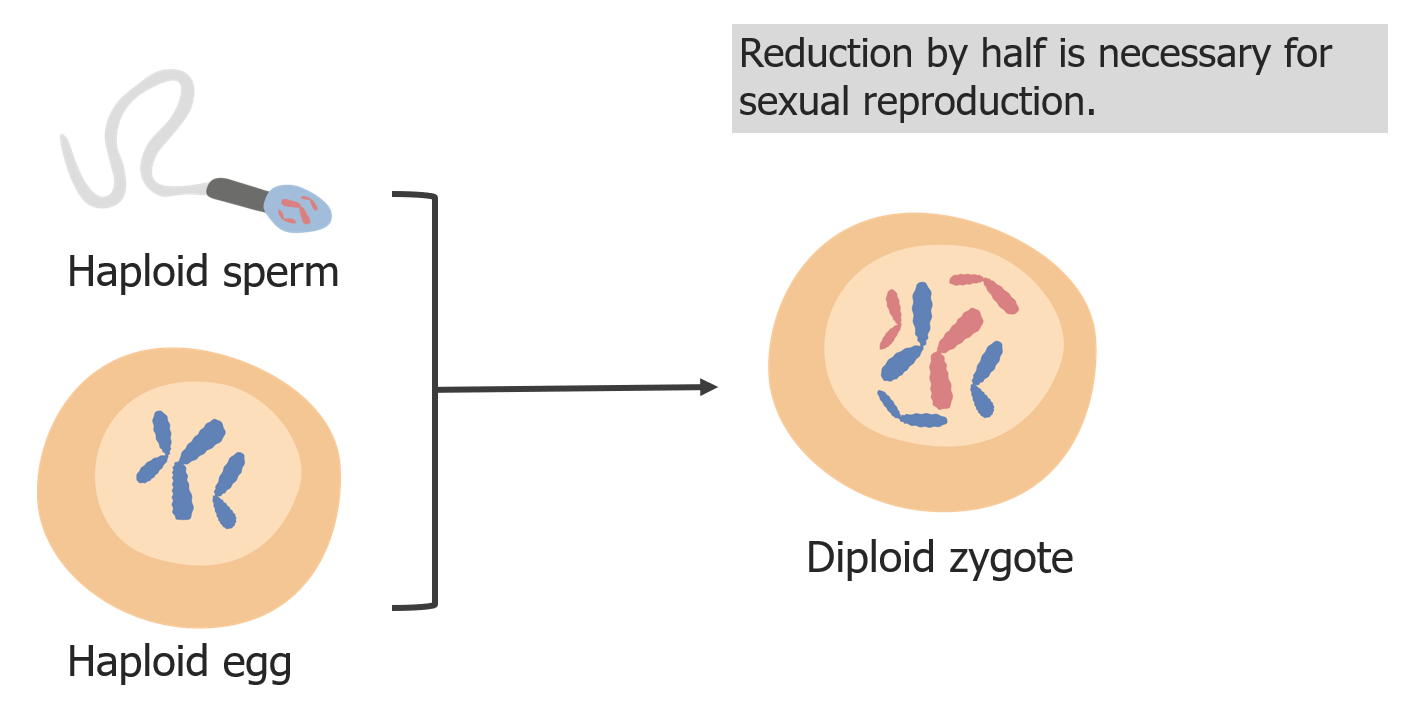
A redução cromossómica é necessária para que o zigoto seja diplóide na fertilização.
Imagem por Lecturio.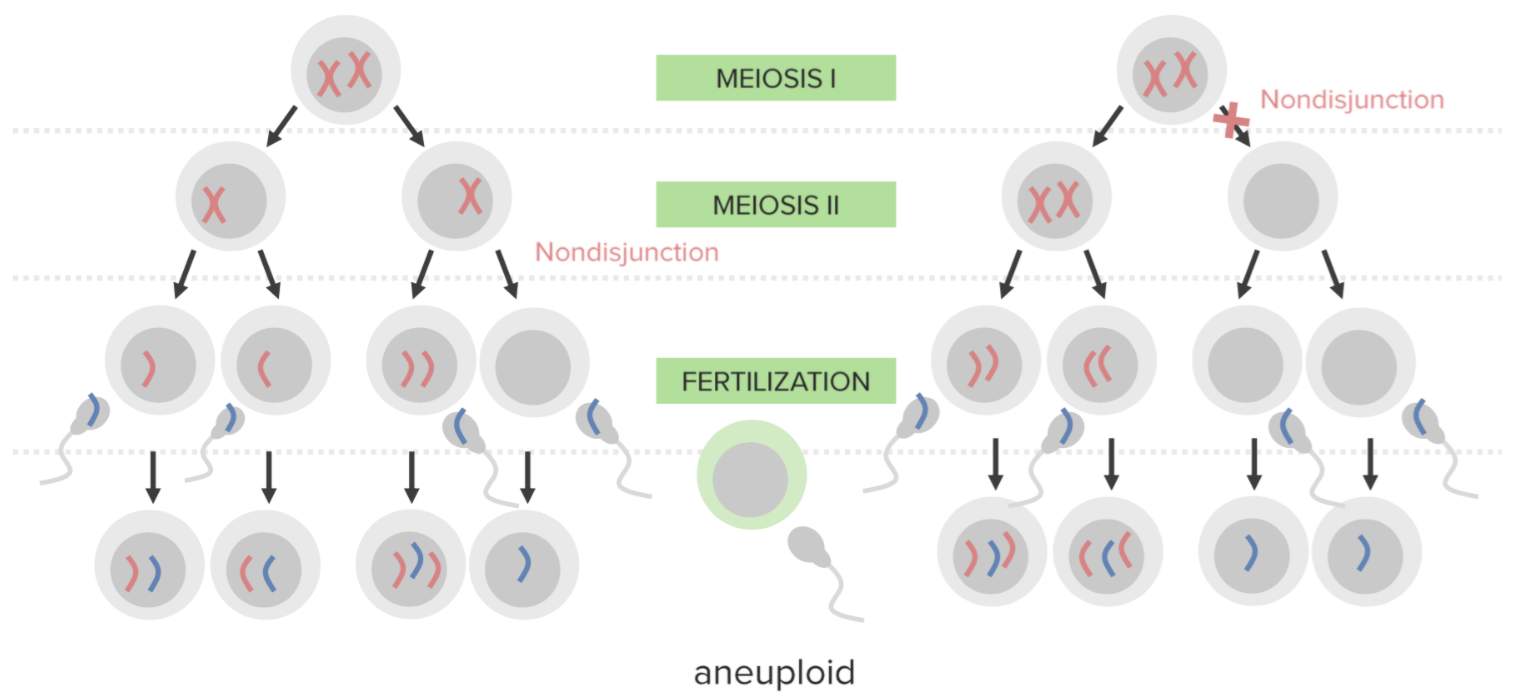
Não disjunção:
A falha em separar adequadamente 2 cromossomas homólogos ou cromatídeos irmãos durante a divisão celular. A não disjunção resulta em aneuploidia (um estado de desequilíbrio cromossómico).

Um diagrama que ilustra o mecanismo etiológico da não disjunção na trissomia:
Um óvulo portador de 2 cópias do mesmo cromossoma adquire outro quando fertilizado, resultando em 3 cópias.
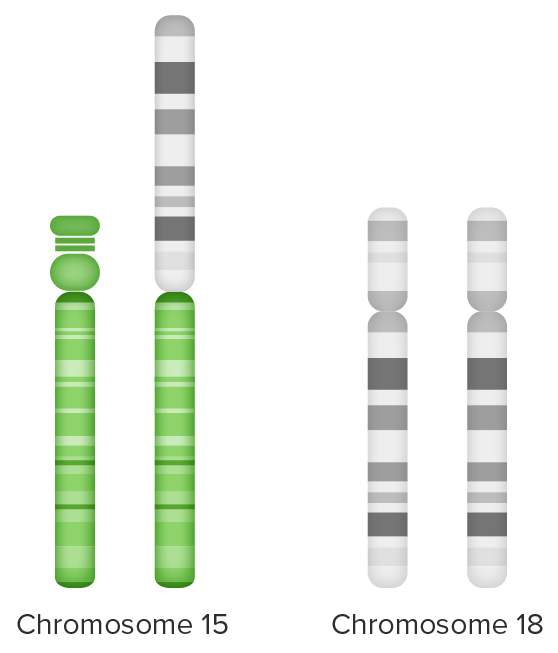
Translocação desequilibrada:
Material extra do cromossoma 18 (braço longo) ligado ao cromossoma 15 (à esquerda) e
trissomia parcial 18 de 2 conjuntos de cromossomas 18 + cromossoma de braço extra longo 18 (à direita)
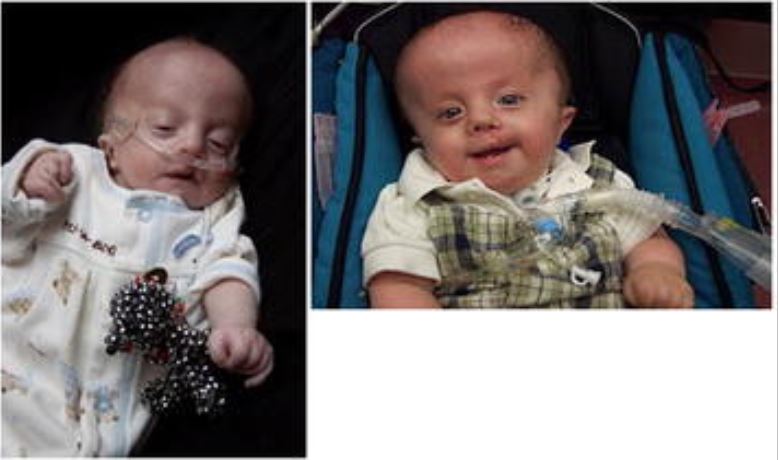
Rapaz com trissomia 18 como lactente e com 1 ano:
Observe a característica da mão de dedos sobrepostos e a colocação de traqueostomia.
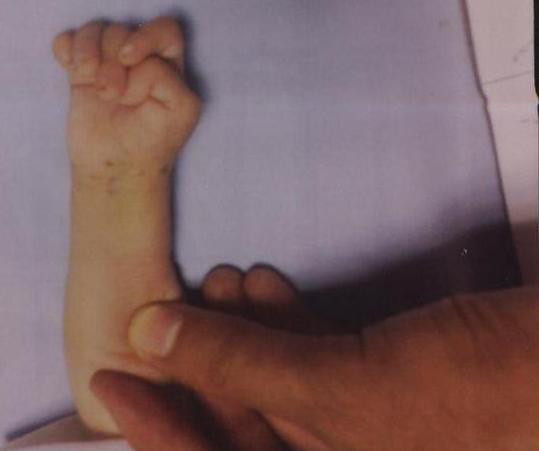
Lactente com trissomia 18 e dedos sobrepostos
Imagem: “Congenital hydrocephalus in an Egyptian baby with trisomy 18: a case report” por Metwalley KA, Farghalley HS, Abd-Elsayed AA. Licença: CC BY 2.0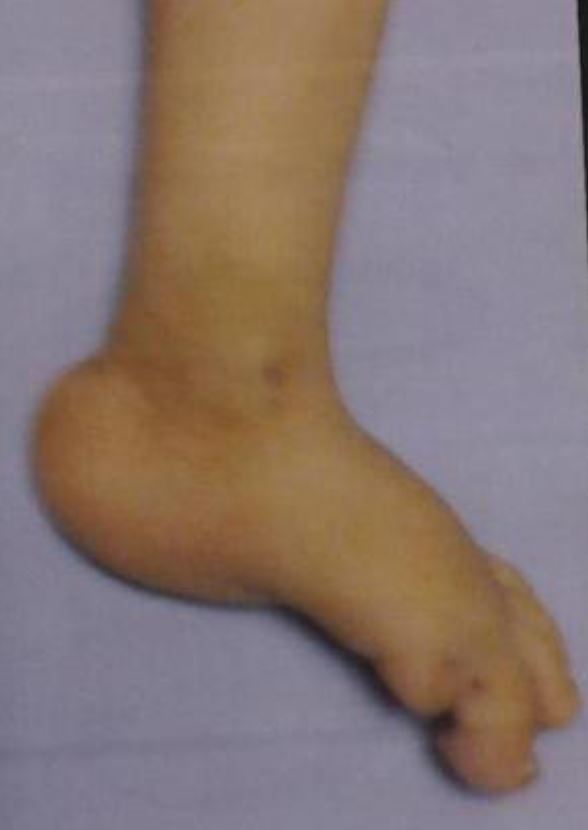
Lactente com trissomia 28 e pé em rotação externa — “Rocker-bottom feet” (um achado comum)
Imagem: “Congenital hydrocephalus in an Egyptian baby with trisomy 18: a case report” por Metwalley KA, Farghalley HS, Abd-Elsayed AA. Licença: CC BY 2.0| 1º trimestre | 2º trimestre | ||||||
|---|---|---|---|---|---|---|---|
| TN | PAPP-A | hCG | AFP AFP The first alpha-globulins to appear in mammalian sera during fetal development and the dominant serum proteins in early embryonic life. Hepatocellular Carcinoma (HCC) and Liver Metastases | Estriol Estriol A hydroxylated metabolite of estradiol or estrogen that has a hydroxyl group at C3, 16-alpha, and 17-beta position. Estriol is a major urinary estrogen. During pregnancy, a large amount of estriol is produced by the placenta. Isomers with inversion of the hydroxyl group or groups are called epiestriol. Noncontraceptive Estrogen and Progestins | hCG | Inhibina A | |
| Trissomia 13 | ↑ | ↓↓ | ↓ | Inalterado | Inalterado | Inalterado | Inalterado |
| Trissomia 18 | ↑↑ | ↓↓ | ↓↓ | ↓ | ↓↓ | ↓↓ | Inalterado |
| Trissomia 21 | ↑↑ | ↓↓ | ↑ | ↓ | ↓ | ↑ | ↑ |
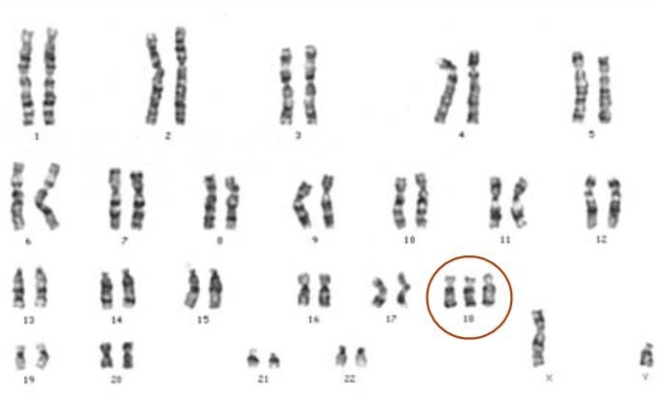
Cariótipo de um indivíduo com síndrome de Edward:
Pode ver-se três cópias do cromossoma 18
