


A síndrome de Chédiak-Higashi (SCH) é uma doença autossómica recessiva causada por mutações que afetam uma proteína reguladora do tráfego lisossómico. Esta proteína desempenha um papel crucial na incapacidade dos neutrófilos de matar micróbios fagocitados. Doentes com SCH apresentam infeções piogénicas recorrentes, hemorragia e hematomas fáceis, e manifestações neurológicas. A síndrome também está associada ao albinismo oculocutâneo. O diagnóstico é feito com base na análise do esfregaço de sangue ou medula óssea do doente e testes Testes Gonadal Hormones genéticos. O tratamento de escolha é o transplante alogénico de células hematopoiéticas.
Last updated: Dec 15, 2025
Sinais e sintomas que geralmente aparecem logo após o nascimento incluem:
Manifestações neurológicas de início tardio que surgem em indivíduos que sobrevivem à primeira infância com tratamento curativo:
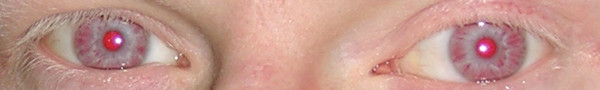
Olhos e cílios hipopigmentados em doente com albinismo oculocutâneo:
pode ser observado na síndrome de Chédiak-Higashi.
A síndrome de Chédiak-Higashi (SCH) pode progredir para a fase acelerada caracterizada por linfohistiocitose hemofagocítica (LHF), onde os leucócitos defeituosos se dividem incontrolavelmente e sofrem metástase. Os sinais e sintomas incluem:
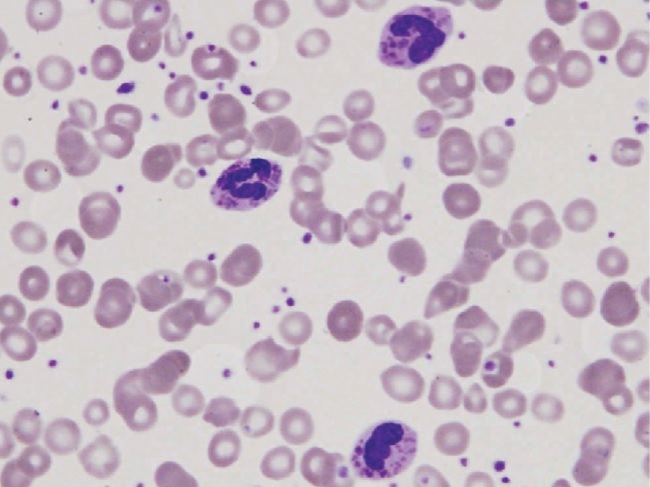
Esfregaço de sangue periférico em doente com síndrome de Chédiak-Higashi que mostra 3 leucócitos polimorfonucleares com grandes grânulos. Estes achados são típicos.
Imagem : “Peripheral blood smear of our patient with Chediak-Higashi” por Morrone K. Licença: CC BY 3.0
Amostras de haste de cabelo sob microscopia de luz de alta potência:
(a): amostra de cabelo de controlo afro-americano, que demonstra pigmento distribuído uniformemente na haste do cabelo
(b): amostra de cabelo de um doente gravemente afetado com síndrome de Chédiak-Higashi que demonstra uma distribuição granular atípica de “grumos pigmentados” na haste capilar
(c): amostra de cabelo de um doente afro-americano com síndrome de Chédiak-Higashi que demonstra um padrão de pigmentação granular atípico semelhante
