El síndrome de Chédiak-Higashi es un trastorno autosómico recesivo causado por mutaciones que afectan a una proteína reguladora del tráfico lisosómico. Esta proteína desempeña un papel crucial en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la incapacidad de los LOS Neisseria neutrófilos para eliminar los LOS Neisseria microbios fagocitados. Los LOS Neisseria pacientes con síndrome de Chédiak-Higashi presentan infecciones piógenas recurrentes, hemorragias y hematomas fáciles y manifestaciones neurológicas. El síndrome también está asociado al AL Amyloidosis albinismo oculocutáneo. El diagnóstico se realiza a partir de análisis del frotis de sangre o de médula ósea del paciente y de pruebas genéticas. El tratamiento de elección es el trasplante alogénico de células hematopoyéticas.
Last updated: Dec 15, 2025
Los LOS Neisseria signos y síntomas que suelen aparecer poco después del nacimiento son los LOS Neisseria siguientes:
Manifestaciones neurológicas de aparición tardía, las cuales aparecen en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum individuos que sobreviven la primera infancia tras tratamiento curativo:
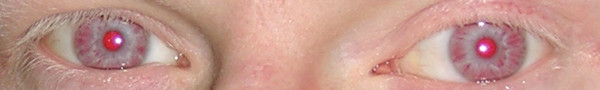
Ojos y pestañas hipopigmentados en un paciente con albinismo oculocutáneo:
Esto puede observarse en el síndrome de Chédiak-Higashi.
El síndrome de Chédiak-Higashi puede progresar a una fase acelerada caracterizada por linfohistiocitosis hemofagocítica (LHH), en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la que leucocitos hiperactivos causan inflamación excesiva y daño tisular. Se desarrolla en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el 80-85% de los LOS Neisseria pacientes con síndrome de Chédiak-Higashi y suele ser letal. Los LOS Neisseria signos y síntomas incluyen:
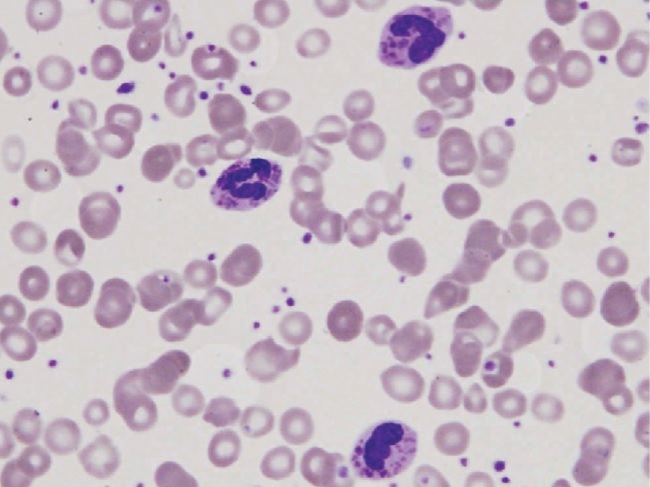
Frotis de sangre periférica en un paciente con síndrome de Chédiak-Higashi que muestra 3 leucocitos polimorfonucleares con gránulos grandes. Estos resultados son típicos.
Imagen: “Peripheral blood smear of our patient with Chediak-Higashi” por Morrone K. Licencia: CC BY 3.0
Muestras del tallo del cabello bajo microscopio de luz de alta potencia:
(a): Muestra de cabello afroamericano de control, que muestra una distribución uniforme del pigmento en el tallo del cabello
(b): muestra de cabello de un paciente con síndrome de Chédiak-Higashi severamente afectado que muestra una distribución granular atípica de “grumos pigmentados” en el tallo del cabello
(c): muestra de cabello de un paciente afroamericano con síndrome de Chédiak-Higashi que muestra un patrón de pigmentación granular atípico similar