


O retinoblastoma Retinoblastoma Retinoblastoma is a rare tumor but the most common primary intraocular malignancy of childhood. It is believed that the condition arises from a neuronal progenitor cell. Retinoblastoma can be heritable or non-heritable. Retinoblastoma embora seja um tumor Tumor Inflammation raro, é o tumor Tumor Inflammation maligno primário do globo ocular mais MAIS Androgen Insensitivity Syndrome frequente em crianças. Pensa-se que a doença surge a partir de uma célula neuronal progenitora. O retinoblastoma Retinoblastoma Retinoblastoma is a rare tumor but the most common primary intraocular malignancy of childhood. It is believed that the condition arises from a neuronal progenitor cell. Retinoblastoma can be heritable or non-heritable. Retinoblastoma pode ser hereditário ou não hereditário. A doença manifesta-se tipicamente pela presença de leucocória unilateral ou bilateral (reflexo branco anormal no olho) numa criança com menos de 2 anos. O retinoblastoma Retinoblastoma Retinoblastoma is a rare tumor but the most common primary intraocular malignancy of childhood. It is believed that the condition arises from a neuronal progenitor cell. Retinoblastoma can be heritable or non-heritable. Retinoblastoma é uma doença potencialmente mortal se não for tratada, mas um diagnóstico precoce está associado a elevadas taxas de sobrevivência em países desenvolvidos.
Last updated: Dec 15, 2025
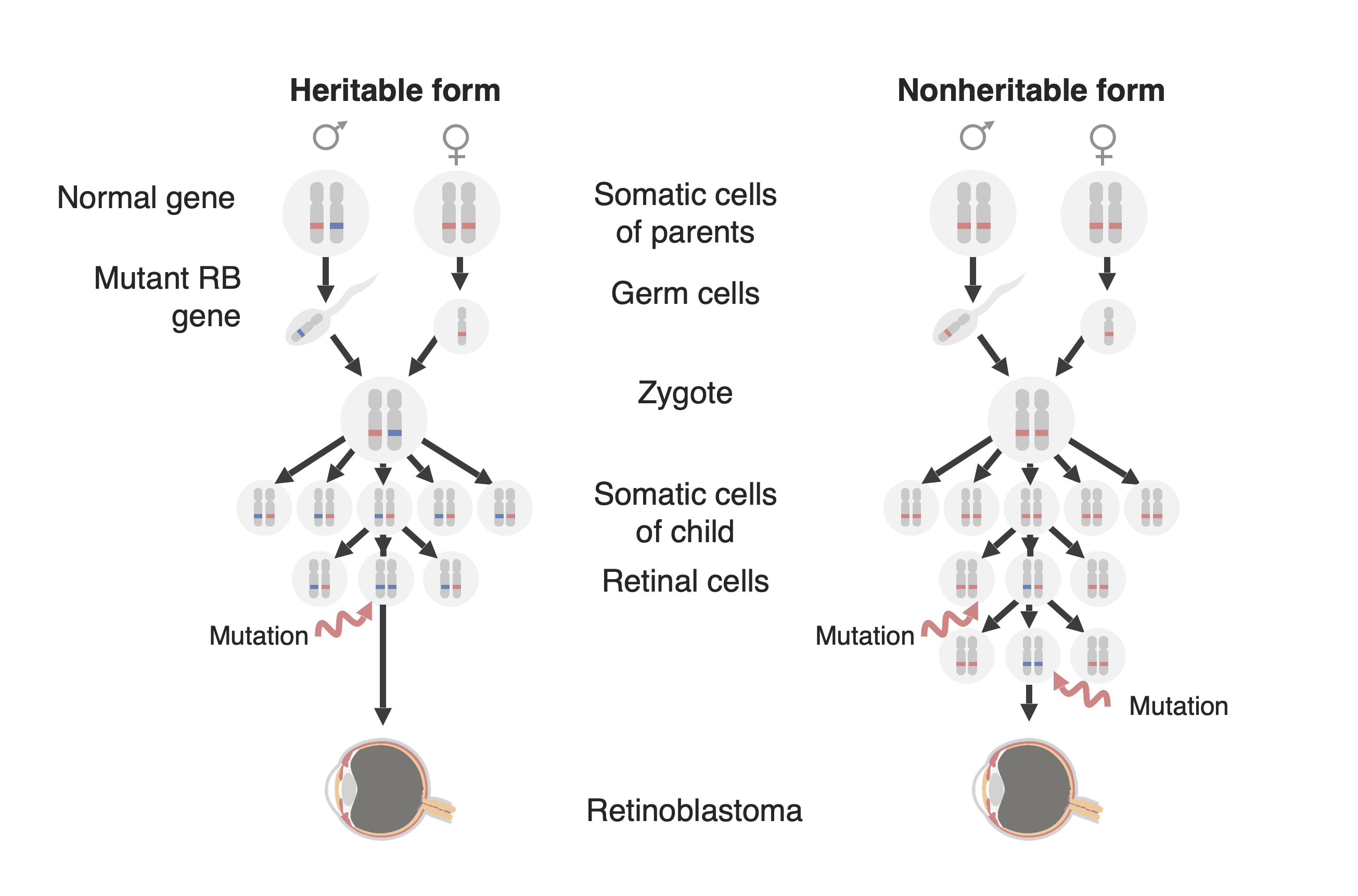
Diagrama das duas formas de desenvolvimento de retinoblastoma: à esquerda a forma familiar ou forma hereditária e à direita a forma esporádica ou forma não hereditária. Na forma hereditária, um gene RB1 mutado (em azul) é transmitido à criança e apenas é necessária uma mutação adicional no alelo normal (em vermelho) para o desenvolvimento do tumor. Na forma não hereditária, a mutação do gene RB1 pode surgir na linhagem germinativa (não mostrada nesta figura) comportando-se como a forma hereditária ou, uma célula precursora da retina pode adquirir uma mutação RB1 em ambos os alelos e desenvolver-se um retinoblastoma.
Imagem de Lecturio.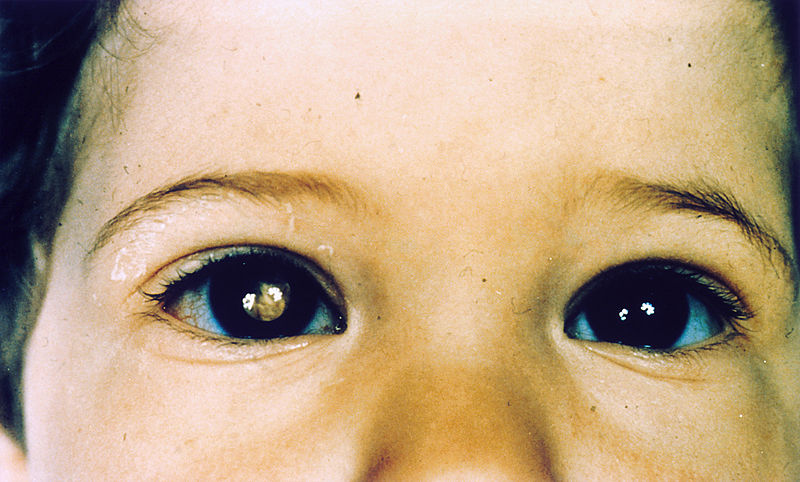
Uma criança com retinoblastoma no olho direito com leucocória
Imagem: “Pathology: Patient: Retinoblastoma” por The National Cancer Institute. Licença: Public Domain
Criança que apresenta celulite orbitária do olho direito devido a um retinoblastoma localmente avançado
Imagem: “Orbital cellulitis” por The Pan African Medical Journal. Licença: CC BY 2.0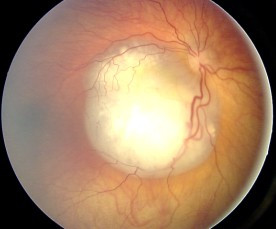
Aspeto do fundo ocular no retinoblastoma
Imagem: “Fundus retinoblastoma” por Aerts, I, Lumbroso-Le Rouic. Licença: CC BY 2.0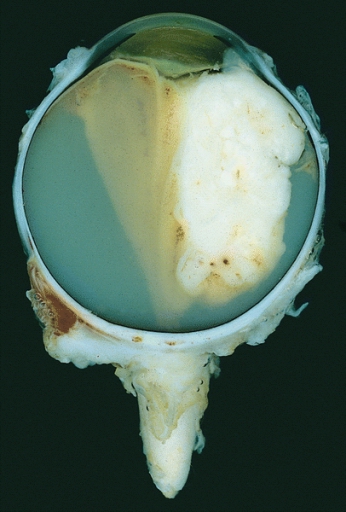
Olho enucleado com um grande retinoblastoma exofítico
Imagem: “Retinoblastoma” por The Armed Forces Institute of Pathology (AFIP). Licença: Public Domain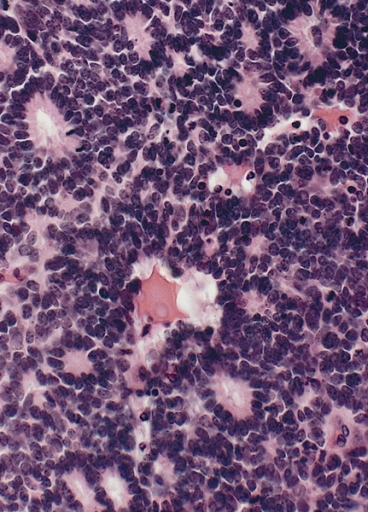
Retinoblastoma com as características rosetas de Flexner-Wintersteiner, com alinhamento circular de células colunares curtas à volta de um lúmen central.
Imagem: “Flexner-Wintersteiner rosettes” por The Armed Forces Institute of Pathology (AFIP). Licença: Public DomainO tratamento depende do estadiamento e existem várias opções terapêuticas disponíveis que permitem a preservação do globo ocular
O diagnóstico diferencial inclui qualquer condição que possa causar leucocória.
