


O raquitismo hipofosfatémico ligado ao X (RHLX) é o mais comum dos vários distúrbios hereditários caracterizados pela excreção de fosfato renal e que leva a ossos frágeis ou moles. Anteriormente conhecido como "raquitismo resistente a vitamina D", o RHLX não é atualmente considerada uma verdadeira resistência à vitamina D (relacionada com defeitos hereditários na via metabólica da vitamina D ou no recetor de calcitriol Calcitriol The physiologically active form of vitamin d. It is formed primarily in the kidney by enzymatic hydroxylation of 25-hydroxycholecalciferol (calcifediol). Its production is stimulated by low blood calcium levels and parathyroid hormone. Calcitriol increases intestinal absorption of calcium and phosphorus, and in concert with parathyroid hormone increases bone resorption. Parathyroid Glands: Anatomy). A apresentação clínica típica inicia-se durante a infância e manifesta-se como baixa estatura, joelhos em valgo ou em varo, dor óssea e odontalgia. O diagnóstico é feito através de estudos laboratoriais e confirmado pela identificação da mutação no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics PHEX PHEX A membrane-bound metalloendopeptidase that may play a role in the degradation or activation of a variety of peptide hormones and intercellular signaling peptides and proteins. Genetic mutations that result in loss of function of this protein are a cause of hypophosphatemic rickets, X-linked dominant. X-linked Hypophosphatemic Rickets. O tratamento inclui burosumab a administração de elevadas doses de vitamina D ( calcitriol Calcitriol The physiologically active form of vitamin d. It is formed primarily in the kidney by enzymatic hydroxylation of 25-hydroxycholecalciferol (calcifediol). Its production is stimulated by low blood calcium levels and parathyroid hormone. Calcitriol increases intestinal absorption of calcium and phosphorus, and in concert with parathyroid hormone increases bone resorption. Parathyroid Glands: Anatomy) e fosfato. Para garantir o crescimento adequado, é fulcral o acompanhamento destes pacientes por uma equipa multidisciplinar.
Last updated: Dec 15, 2025
O raquitismo hipofosfatémico ligado ao X (RHLX) é uma doença genética rara que causa hipofosfatémia e consequente raquitismo clínico.
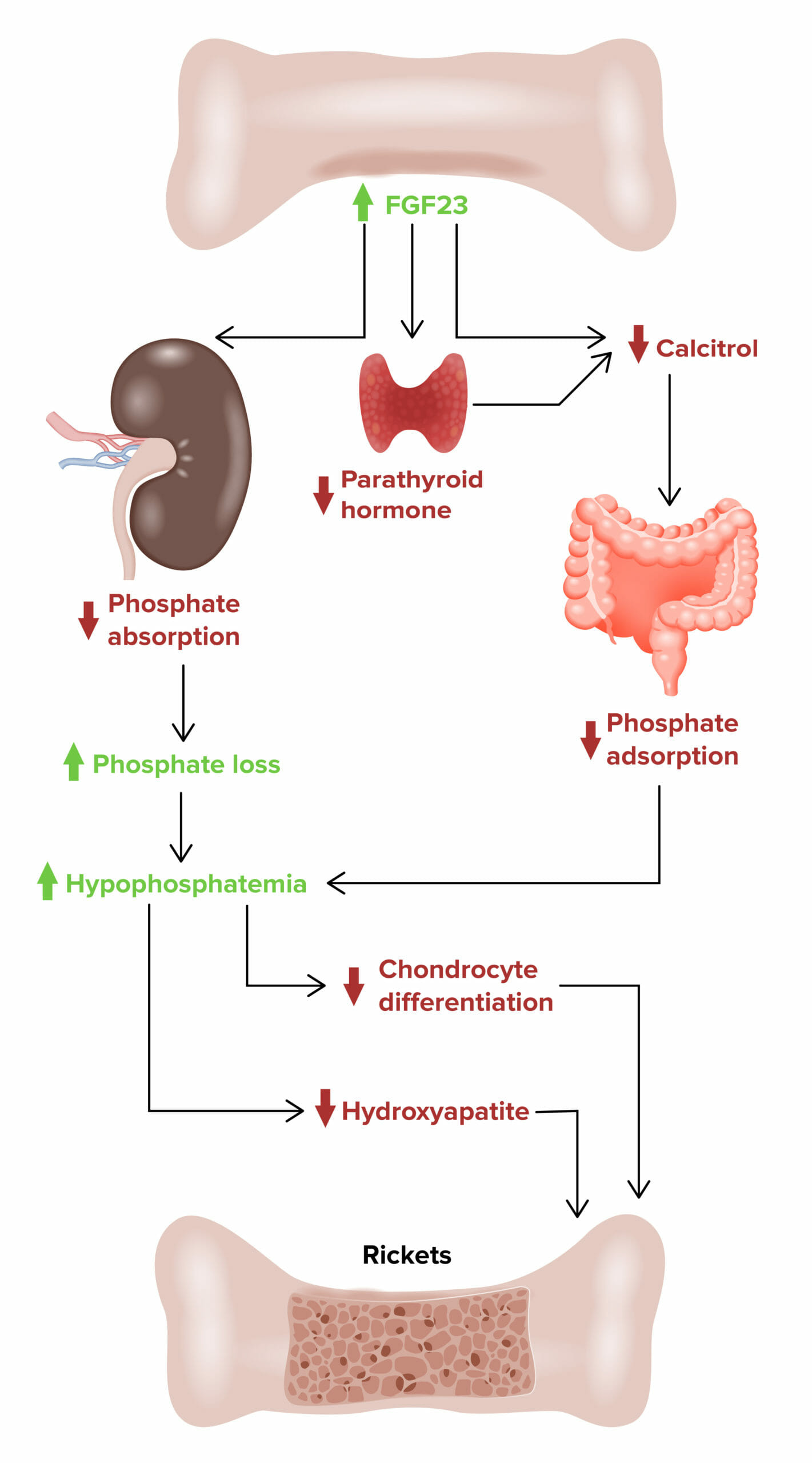
Raquitismo hipofosfémico ligado ao X:
Efeito do fator de crescimento dos fibroblastos 23 (FGF23, pela sigla em inglês) no desenvolvimento de raquitismo hipofosfémico ligado ao X
A apresentação clínica dos RHLX pode variar de assintomática a gravemente sintomática. O raquitismo hipofosfatémico ligado ao X é geralmente confundido com o raquitismo nutricional nos bebés, e os sintomas incluem:
Antecedentes familiares:
Testes Testes Gonadal Hormones de laboratório:
Testes Testes Gonadal Hormones genéticos:
Imagiologia:
Deve ser realizada a avaliação radiológica para excluir a curvatura fisiológica e a displasia óssea. Potenciais achados no raio-X podem incluir:
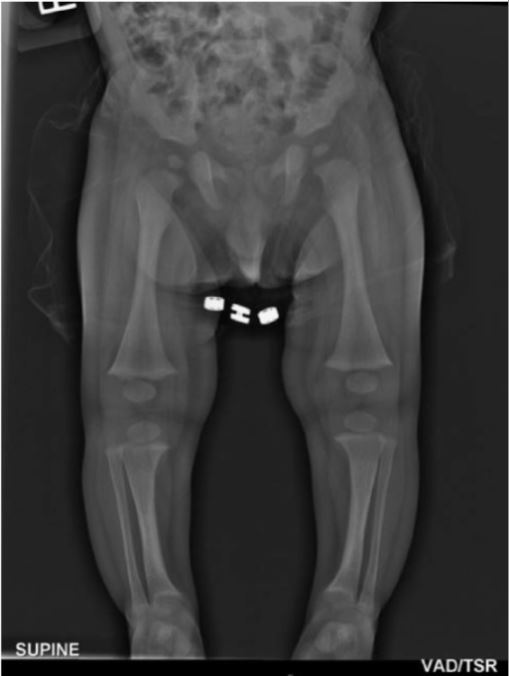
Uma criança de 7 meses com raquitismo hipofosfatémico ligado ao X:
Os ossos longos dos membros inferiores demonstram alargamento das metáfises com desgaste, mas sem alargamento da epífise. Existe também uma ligeira curvatura da tíbia.
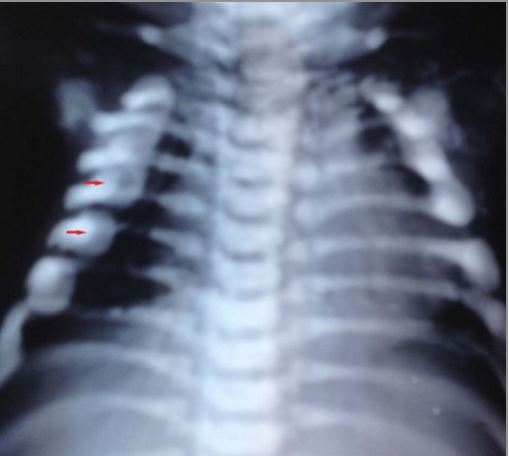
Radiografia que mostra rosário raquítico: poeminências tipo conta nas articulações costocondrais
Imagem: “Malignant infantile osteopetrosis: Case report with review of literature” por Essabar L, Meskini T, Ettair S, Erreimi N, Mouane N. License: CC BY 2.0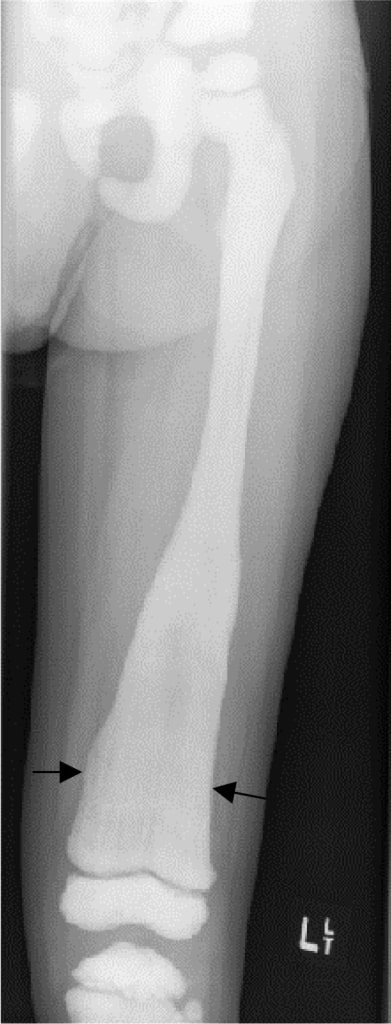
Um raio-X de uma criança de 4 anos com deformidade em frasco de Erlenmeyer do fémur distal, que mostra o reforço da metáfise no raquitismo hipofosfatémico ligado ao X
Imagem: “Osteopetrosis” por Stark Z, Savarirayan R. Licença: CC BY 2.0Os objetivos do tratamento são melhorar a osteomalácia e as deformidades esqueléticas, melhorar o crescimento dos bebés, a atividade física das crianças e diminuir a dor.
Alguns adultos com RHLX podem ter problemas médicos ligeiros, enquanto outros podem apresentar dor persistente ou complicações. É imperativa a monitorização do tratamento para potenciais complicações do hiperparatiroidismo e nefrocalcinose.
